| ORIGINAL ARTICLE |
|
|
1 Institute of Brain Science and Disorders, Qingdao University, Qingdao, China;
2 Brain Function and Disease Laboratory, Shantou University Medical College, Xin-Ling Road #22, Shantou 515041, China
Corresponding Author: Jie Wu, MD, PhD, Professor, Institute of Brain Science and Disorders, Qingdao University, Qingdao, China; Brain Function and Disease Laboratory, Shantou University Medical College, Xin-Ling Road #22, Shantou 515041, China. E-mail: jiewu2@qdu.edu.cn or jiewubni@gmail.com.
Running title: CB2R-MEDIATED A REDUCTION OF OAβ-INDUCED NEURONAL HYPEREXCITATION
| |
ABSTRACT |
| INTRODUCTION | |
|
|
MATERIALS AND METHODS |
|
|
RESULTS |
|
|
DISCUSSION |
|
|
CONCLUSIONS |
|
|
ACKNOWLEDGMENTS |
|
|
CONFLICTS OF INTEREST |
|
|
AUTHOR CONTRIBUTIONS |
|
|
DATA AVAILABILITY STATEMENT |
|
|
REFERENCES |
|
|
ABSTRACT
|
|---|
There is a significantly elevated incidence of epilepsy in Alzheimer’s disease (AD). Burgeoning evidence indicates that soluble beta-amyloid peptides oligomers (oAβ) are vital players in driving neuronal hyperactivity in AD. It is well known that the modulations of the cannabinoid system exhibit neuroprotective effects in various neurological diseases, including AD. However, a consensus is yet to emerge as to the impact of hippocampal cannabinoid receptor 2 (CB2R) in protecting hippocampal neurons against Aβ-induced neuronal hyperexcitation. Here, we report that chronic treatment of primary hippocampal neuronal cultures with 100 nM Aβ1–42 oligomers for 7 days results in a neuronal hyperexcitation. Further, pre-treatments of CB2R agonist (JWH133, 1 μM with Aβ1–42 for 7 days) significantly protect hippocampal neurons against Aβ-increased hyperexcitation, including prolonged action potential (AP) initiation, enhanced after hyperpolarization (AHP), and decreased AP numbers. These effects are eliminated by a selective CB2R antagonist, AM630. Furthermore, when the oAβ-increased neuronal hyperexcitation has already formed (pretreated with oAβ1–42 for 5 days), the addition of JWH133 also abolishes the Aβ’s effects. Collectively, our results suggest that the selective activation of hippocampal CB2Rs not only prevents Aβ-increased neuronal hyperexcitation, but also abolishes the established neuronal hyperexcitation, which underlies our recent findings that CB2Rs play a critical role in protection of hippocampal neurons against the Aβ-induced neuronal toxicity and degeneration. This novel finding suggests a potentially therapeutic strategy for the treatment of AD using CB2R agonists.
KEY WORDS: Amyloid beta-peptide; Cannabinoid receptor 2; Hippocampal neurons; AD, Alzheimer's disease; Neuronal hyperexcitation; Neuronal toxicity|
|
INTRODUCTION |
|---|
Alzheimer’s disease (AD) is a neurodegenerative disorder characterized by a progressive impairment of memory and other cognitive functions (1). Amyloid-β (Aβ) plaques and neurofibrillary tangles are the main neuropathological hallmarks in AD (37). Emerging evidence indicates that neuronal hyperactivity is a potential key feature of the early phase of AD. In AD patients, the incidence of seizures is significantly higher than that in non-demented elderly controls (2). Moreover, epileptiform activity has been noted in the entorhinal–hippocampal circuit in excessive accumulation of pathogenic Aβ assemblies in animals (27). However, a mechanism framework linking the abnormal accumulation of Aβ peptides to the paroxysm of seizures is elusive.
The amyloid precursor protein (APP) by proteolytic cleavage generates Aβ, a critical molecule that perturbs network activity in AD. In high Aβ plaque burden neurons demonstrate tonic hyperexcitability in multiple models of AD, For example, frontal cortex or hippocampus (8). Accumulating lines of evidence indicate that Aβ oligomers can also induce neuronal hyperexcitability in the hippocampus with the absence of plaque pathology, which suggests that compared to Aβ plaques, soluble Aβoligomers are significant drivers and lead to neuronal dysfunction and degeneration 41).
Endocannabinoid (eCB) system is one of the main neuroregulatory systems acting in the central nervous system (CNS), which controls neuronal activity by activating cannabinoid receptors (CBRs) (21). Identifying regional changes of cannabinoid receptor-1 and receptor-2 (CB1R and CB2R) expression is particularly important when considering endocannabinoid system-based therapies (25). It is well known that CB1Rs are predominantly expressed in the central nervous system, whereas CB2Rs are mainly expressed in peripheral immune cells (11). However, this concept has been challenged. CB2Rs are expressed not only on glial cells but also on the neurons of different brain areas, including hippocampus (29). We recently report that the activation of CB2Rs reduces neuronal firing and excitability in both VTA DA neurons and red nuclear neurons (38, 39). It has also reported that CB2R agonist exhibits its effects in long lasting hyperpolarization and mediation of neuron-autonomous slow self-inhibition (30).
Recently, we found that the activation of CB2Rs protects hippocampal neurons against chronic Aβ-induced toxicity in the primary hippocampal cultures (40), however, the underlying mechanisms are still unclear, and one possibility is the CB2R-mediated a reduction of neuronal hyperexcitation by chronic Aβ treatment. In this study, we tested this possibility by measuring the intrinsic activity to elucidate the protective roles of CB2Rs in Aβ-induced hippocampal neuronal hyperexcitation.
|
|
MATERIALS AND METHODS |
|---|
Preparation of rat hippocampal primary neuron cultures
The protocol for the preparation of neuronal cultures from rodents was approved by the Institutional Animal Care and Use Committee of the Qingdao University. Poly-D-lysine, 0.02 % solution, was added to culture dishes the day before culture. Dishes were swirled to make sure the entire bottom was coated, and then dishes were left in a 37°C/5% CO2 incubator overnight. The next day, dishes were washed three times with sterile water and left in the incubator after the final wash. Postnatal SD rats (P0−1day-old) were sacrificed, and the CA1-CA3 region of the hippocampus was dissected under a stereological microscope. The tissue was minced with scissors in an ice-cold neurobasal medium and then digested with Papain at 30°C for 20 min in tubes shaken at 120 rpm in a water bath shaker. After enzyme digestion, the reaction was stopped by adding inactivated fetal bovine serum into the medium. Then, digested tissue was filtered and transferred into 15 ml tubes. After trituration, tissue was centrifuged at 1500 rpm for 3 min to form pellets containing dissociated cells. The supernatant was removed and replaced with a Neurobasal medium, which was used to resuspend the pellets. This process was repeated 3 times. After the final centrifugation, the supernatant was replaced with a Neurobasal medium supplemented with 0.5 % (w/v) L-glutamine and 2% B27 serum-free supplement. Cells were suspended and counted based on trypan blue exclusion and plated at a density of 1.0 × 106 cells per well in culture dishes. Cells were kept within the 37°C/5% CO2 incubator for future use.
Aβ preparation and treatment
Aβ1–42 peptides were purchased from the MCE Company (Shanghai, China). Based on the introduction of the preparation of the oligomer form of Aβ1–42 peptide, it was dissolved in 1,1,1,3,3,3-hexafluoro-2-propanol at a concentration of 1 mol/L in 2.217 mL aliquots, air-dried in the fume cupboard, and stored at −20°C. The clear film obtained after HFIP volatilizing was resuspended in dimethyl-sulfoxide and was further diluted using PBS (pH 7.4) to a final concentration of 100 μM and incubated at 4°C for 24 h without shaking. Following incubation, centrifuged at 13,000 rpm, for 10 min in the cold, transferred supernatant to a new tube, and the oligomeric Aβ1–42 was prepared.
Primary cultured neurons were maintained at 37°C 5 % CO2 in an incubator for 7–8 days before Aβ exposure. The Aβ-containing culture medium (100 nM) was replenished daily for 7–8 days. The same procedure was followed with medium for the control group, but Aβ exposure was not included. For pharmacological studies, the CB2R agonist (JWH133) or antagonist (AM630) was applied for 40 min before Aβ exposure.
Neuronal viability assay
The neuronal viability assay was performed using a lactate dehydrogenase (LDH) reagent kit. LDH is an oxidoreductase enzyme that catalyzes the interconversion of pyruvate and lactate. LDH is released into the culture supernatant by cells after their membrane is damaged. In a 96-well plate, 5 μL of culture supernatant was added into duplicate wells, and the samples were brought to a final volume of 50 μL with LDH Assay Buffer. Then, 50 μL of the Master Reaction Mix was added to each of the wells. The plate was incubated at 37°C. The change is calculated in measurement using “Initial” to “Final” from the samples.
Electrophysiology
Patch-clamp recordings in cultured hippocampal neurons were performed. The standard external solution contained 140 mM NaCl, 3 mM KCl, 2 mM MgCl2, 2 mM CaCl2, 10 mM D-glucose, 10 mM HEPES, pH 7.4 with Tris base. The pipette solution for current-clamp recordings was used to measure intrinsic excitability contained 130 mM KMeSO4, 10 mM KCl, 10 mM HEPES/K-HEPES, 2 mM MgSO4, 0.5 mM EGTA, and 3 mM ATP, pH 7.3 with KOH. Recordings were considered acceptable only if cells exhibited overshooting action potentials (AP) and their resting membrane potential did not vary by>5 mV. Neuronal intrinsic excitability was measured as the number of AP spikes in response to a series of fixed, 2, or 4 s current injection steps (60, 90, 120, or 150 pA). Data were acquired at 2 kHz using an Axopatch 200B amplifier and analyzed using clampfit 10.
Data analysis and statistics
Data are presented as mean ± SEM with the number of samples (n). A probability level of p<0.05 is considered to be statistically significant. Significant differences were determined using the two-tailed Student’s-test or one-way ANOVA as appropriate.
|
|
RESULTS |
|---|
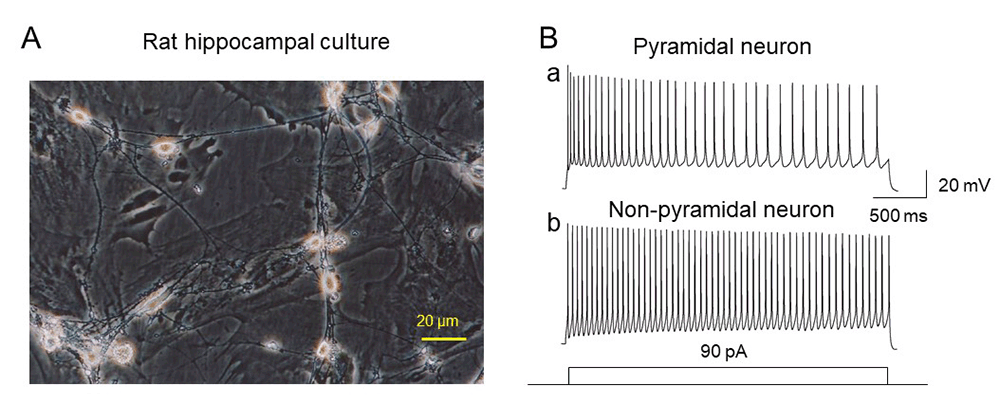
Identification of cell phenotype
Pyramidal cells in rats’ primary hippocampal cultures were initially and tentatively identified based on their morphology (Fig. 1A) and the characterizations based on their electrophysiological features. Figure 1Ba and b compared neuronal AP firing rates between pyramidal (Ba) and nonpyramidal neurons (Bb) upon injection of 90 pA depolarizing current. In 20 cells tested, the averaged AP firing rate for pyramidal neurons was 15.9 ± 0.8 Hz (n=12), while that for nonpyramidal neurons was 29.8 ± 1.3 Hz (n=8, pyramidal vs. nonpyramidal, T-test, p<0.001) induced by the injection of 90 pA depolarizing current. In the following experiments, we used pyramidal neurons as experimental preparation.
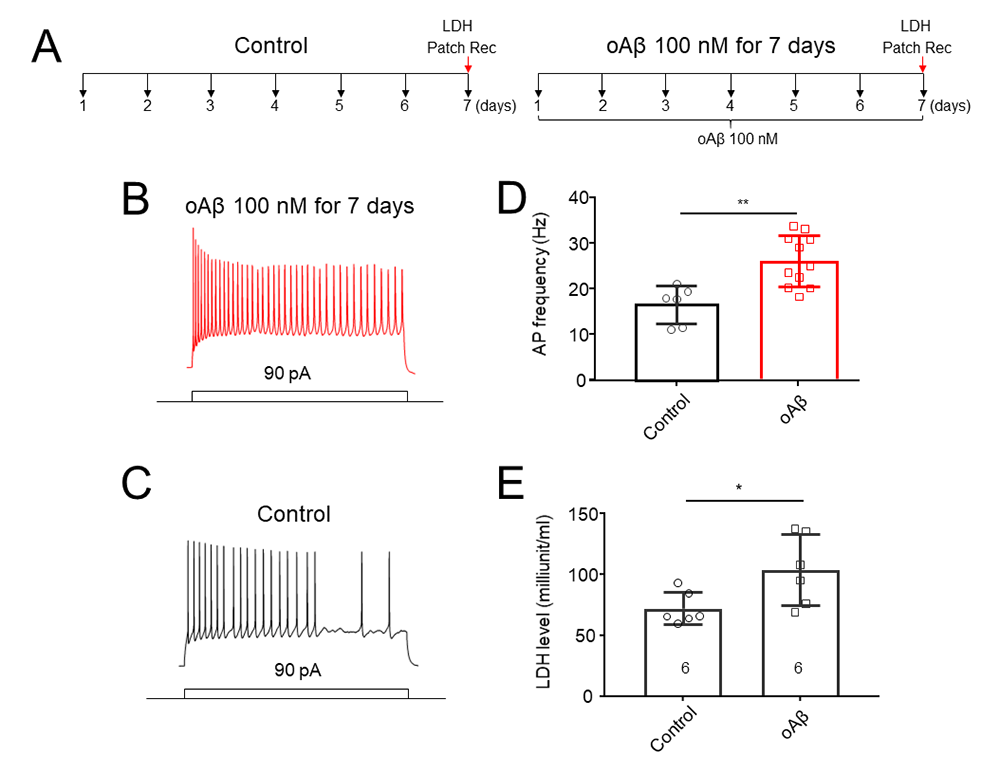
Chronic oligomeric Aβ1–42 induced neuronal hyperexcitation and toxicity in hippocampal cultures
Initial experiments were designed to confirm the chronic Aβ1-42 oligomers (oAβ)-induced neuronal hyperexcitation and toxicity. As showed in experimental protocol (Fig. 2A), after exposed to 100 nM oAβ for 7 days, cultured hippocampal pyramidal neurons exhibited an increased AP firing rate (Fig. 2B) after current injection (90 pA) compared to control cells (Fig. 2C). Statistical analysis showed that chronic oAβ treatment significantly increased neuronal AP firing rate (Fig. 2D). Then, we measured LDH release in two experimental groups: control (untreated) and oAβ treated groups. The results showed that the oAβ induced an increase in LDH level compared to control group, suggesting that chronic treatment with oAβ results in a neuronal toxicity in primary hippocampal cultures (Fig. 2E).
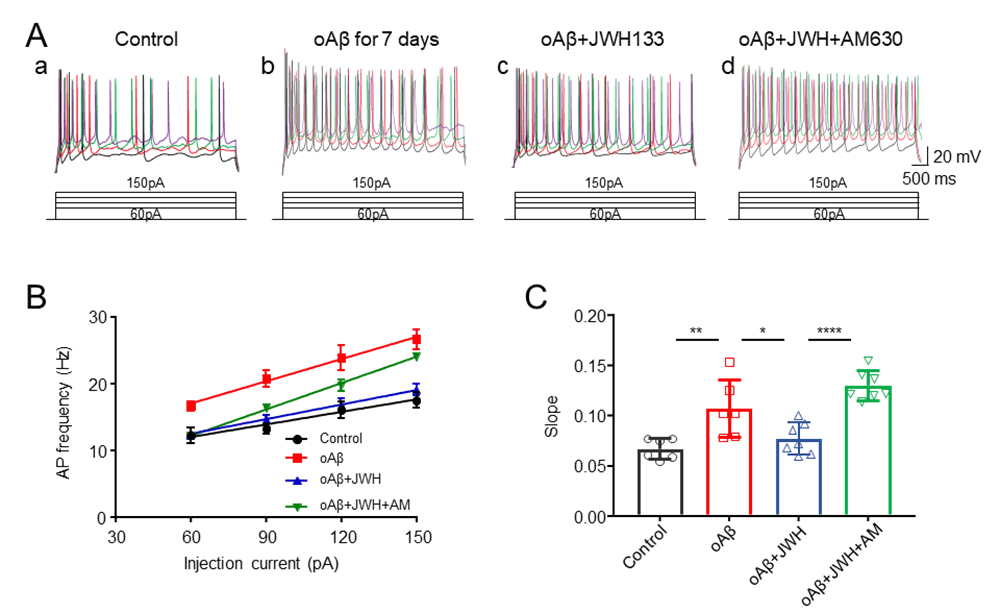
Effects of activation of CB2R on the chronic oAβ treatment-induced neuronal hyperexcitation
Experiments were classified into 4 groups: control, oAβ, oAβ + JWH133 (CB2R agonist), and oAβ + JWH133 + AM630 (CB2R antagonist). In oAβ treated group, hippocampal neuronal cultures were treated with oAβ 100 nM for 7 days (change culture medium contained oAβ 100 nM daily). In oAβ + JWH133 group, 1 µM JWH133 was pretreated to cells for 50 min, then, 100 nM oAβ was added. In oAβ + JWH133 + AM630 group, 1 µM JWH133 and 1 µM AM630 were pretreated to cells for 50 min, then, 100 nM oAβ was added. To quantify the changes of neuronal excitability of each experimental group, we injected depolarizing currents (60, 90, 120, 150 pA, respectively) and measured the changes of AP firing rates (Fig. 3Aa-d). The summarized data illustrated that oAβ treatment induced neuronal hyperexcitation, but JWH133 significantly shifted the input-output relationship curve to the right compared with oAβ group alone (Fig. 3B). Statistical comparisons of the slope of AP firing with different injected currents showed that chronic treatment with oAβ induced the hyperexcitation in cultured hippocampal neurons, and the CB2R agonist JWH133 protected neurons against the oAβ-induced hyperexcitation, which is eliminated bya selective CB2R antagonist, AM630 (Fig. 3C). These results suggest that JWH133 eliminates the oAβ-induced neuronal hyperexcitation through the activation of hippocampal CB2Rs.
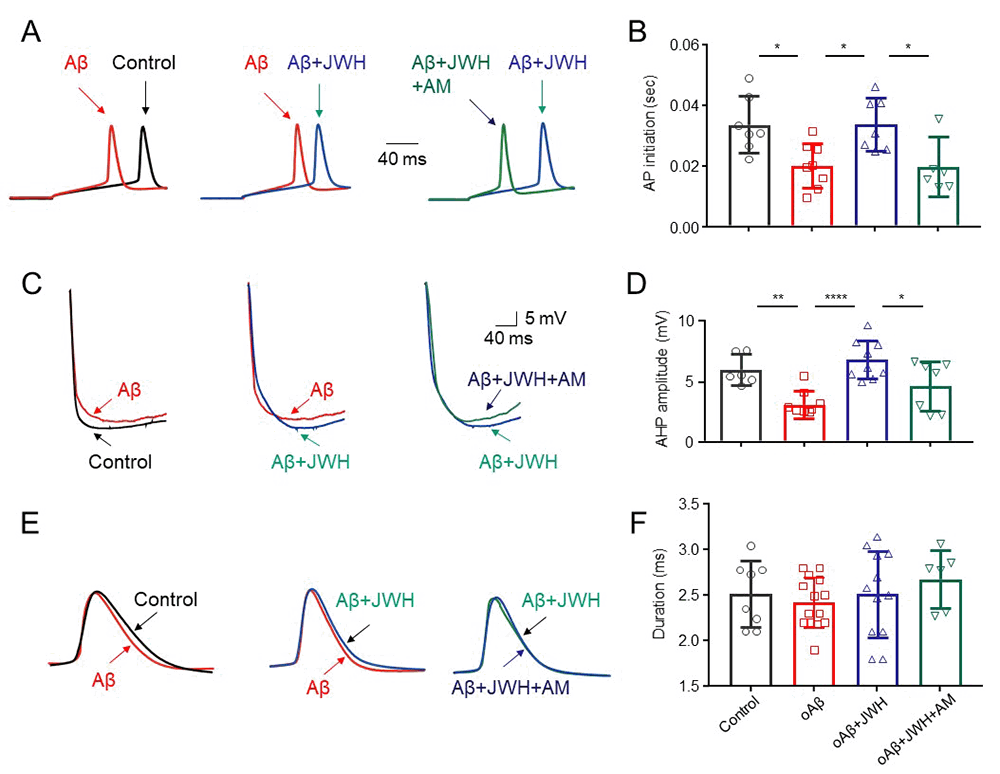
Intrinsic mechanisms of CB2R-mediated reduction of the oAβ-induced hyperexcitation
To elucidate possible intrinsic mechanisms of the CB2R-mediated reduction on the oAβ induced neuronal hyperexcitation, we measured AP initiation latency, AP after hyperpolarization potential (AHP), and AP duration in four experimental groups: control (untreated), oAβ, oAβ + JWH133 (1 μM) and oAβ + JWH133 + AM630 (1 μM). Using an injected current (90 pA) protocol, the initiation latency of APs in oAβ treated group was shorter than that in control group. However, with oAβ and JWH133 co-treatment, the oAβ’s effect was eliminated. When neurons were co-treated with oAβ, JWH133, and AM630, the effect of JWH133 can be prevented (Fig. 4A, B). Using the same experimental protocol, we further measured neuronal AP, AHP and duration in the above 4 groups, the results showed that the oAβ treatment reduced AHP level (more depolarization), while JWH133 enhanced the AHP level, and the JWH133’s effect can be eliminated by AM630 (Fig. 4C, D). However, there were no significant difference of AP duration among the 4 groups (Fig. 4E, F). These results suggest that the activation of CB2Rs prolongs AP initiation latency and increases AHP level, which underlies the CB2R-mediated neuronal protection against chronic oAβinduced neuronal hyperexcitation.
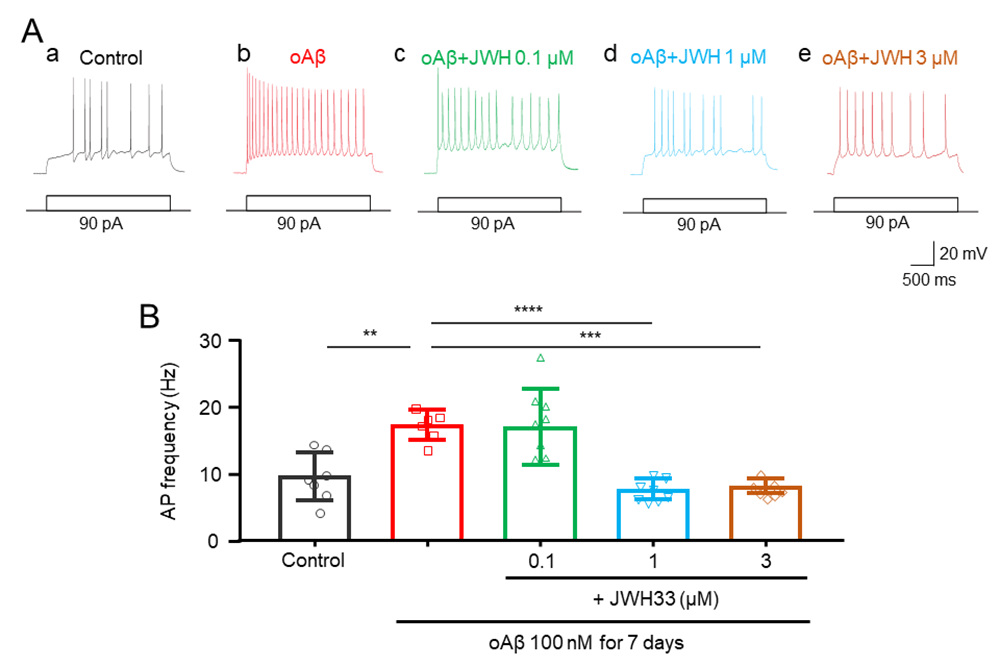
Concentration-effect relationship of JWH133 on the Aβ-induced neuronal excitability
To define concentration-dependent effects of treatment with JWH133 on the oAβ-induced hippocampal pyramidal neuronal hyperexcitation, we treated these cultures using oAβ (100 nM) with 0.1, 1, and 3 μM JWH133, respectively. The results showed that in the 90 pA current injection protocol, JWH133 reduced oAβinduced neuronal hyperexcitation in a concentration-dependent manner(Fig. 5A, B).
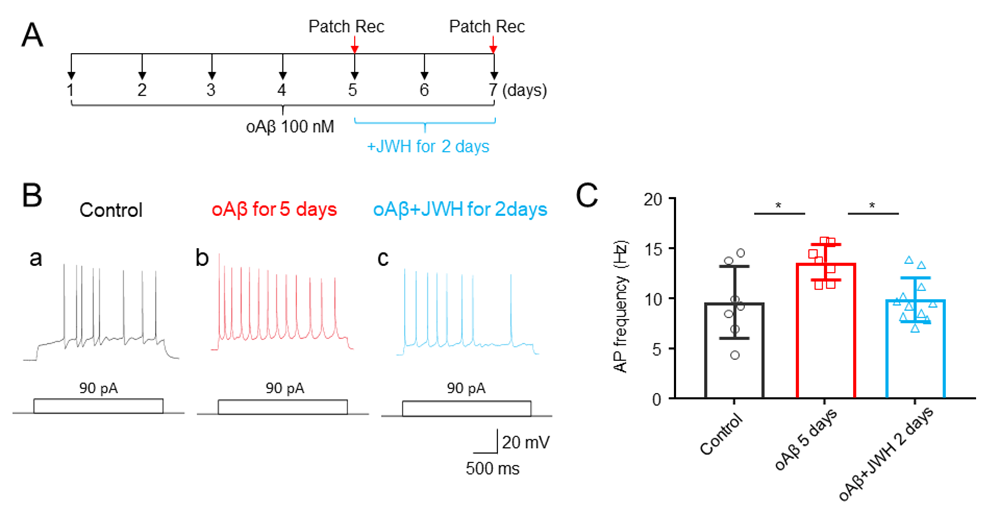
JWH133reduced the oAβ-induced neuronal hyperexcitation expression
Data presented thus far clearly show that co-treatment with JWH133 and oAβ for 7 days prevents the oAβ-induced neuronal hyperexcitation. It is interesting in seeing whether JWH133 can reduce the oAβ-induced neuronal hyperexcitation after its expression. To address this question, we designed an experimental protocol to establish neuronal hyperexcitation after oAβ treatment alone for 5 days, then co-treatment with oAβ1–42 and JWH133 for 2 additional days, and examined whether JWH133 could reduce the established neuronal hyperexcitation by 5-day‘s oAβ1–42 treatment (Fig. 6A). We measured the changes of AP firing rates in 3 experimental groups (Fig. 6): control (untreated), oAβ5days, oAβ5days+ JWH1332days. In the oAβ5days group, hippocampal neuronal cultures were treated with oAβ 100 nM for 5 days and measured neuronal hyperexcitation using patch recordings. In Aβ5days+ JWH1332days group, cultured neurons were exposed to oAβ 100 nM for 5 days, and then JWH133 1 mM was added for following two days. Results showed that compared to the control group, the oAβ5days group induced neuronal hyperexcitation, suggesting that 5-day oAβ 100 nM treatment is enough to induce neuronal hyperexcitation, and this established neuronal hyperexcitation can be eliminated by following two-day’s co-treatment with JWH133. These findings suggest that the activation of CB2Rs can not only prevent the oAβ-induced neuronal hyperexcitation initiation, but also abolish the established neuronal hyperexcitation expression in cultured hippocampal neurons.
|
|
|
|
|
|
|
|
DISCUSSION |
|---|
The major new finding of this study is that a selective activation of hippocampal CB2Rs protects these neurons against chronic oAβinduced neuronal hyperexcitation. We firstly demonstrated that the chronic treatment of rat hippocampal primary cultures with 100 nM oAβ for 7 days enhanced the levels of LDH, which is consistent with our previous findings (20, 40). Then, we showed that the same chronic treatment of oAβ induced a neuronal hyperexcitation, suggesting a success of a cell model of chronic oAβ toxicity/hyperexcitation in hippocampal primary cultures. We used this cell model to evaluate the effects of CB2R agonist on the initiation and the expression of the oAβinduced hippocampal neuronal hyperexcitation. We find that the CB2R agonist, JWH133 not only prevents the oAβ -induced neuronal hyperexcitation, but also eliminates the oAβ -established hyperexcitation. The effects of JWH133 can be abolished by application of AM630, a selective CB2R antagonist, suggesting that the neuroprotective effect of JWH133 is mediated through the neuronal CB2Rs of hippocampal cultures. Collectively, our findings provide the new insights into an understanding of CB2R-mediated neuronal protection for AD prevention and therapeutics.
AD is neurodegenerative dementia characterized by increased accumulation of Aβ, gradual degeneration of neurons of the CNS, and progressive deficits in learning and memory (28)USA. In recent years, studies have shown that a substantial proportion of individuals with AD exhibit epileptiform and seizure activity that exceeds normal population levels (13). Seizures in AD can emerge several years before cognitive symptoms or decline and may persist into the dementia phase, which may underlie the behavioral symptoms and cognitive deficits (33). Meanwhile, emerging evidence indicates that soluble Aβ oligomers are vital players in driving neuronal hyperactivity in AD (7). But the mechanisms underlying this process are still unclear. Therefore, a better understanding of such mechanisms is likely to help improve AD diagnosis and treatment. Recently, we have established a cellular model of Aβ toxicity by a chronic exposure to Aβ1–42 aggregates in rodent hippocampal primary cultures, in which, chronic Aβ1–42 aggregates-induced neuronal hyperexcitation and toxicity (20). We have used this cell model to evaluate the protective effects of CB2Rs on the oAβinduced neuronal toxicity and find that the activation of CB2Rs significantly protects hippocampal neurons against chronic oAβinduced neuronal toxicity (40). However, whether CB2Rs protect hippocampal neurons against the oAβinduced neuronal hyperexcitation remains elusive. In this study, we address this question by using the same cell model and experimental protocol to treat rat hippocampal cultures with Aβ1–42 (oligomers, 100 nM for 7 days), and we confirm that the chronic oAβ treatment induces both neuronal degeneration (e.g., increased LDH level) and hyperexcitation. Then we use this cell model of Aβ toxicity to evaluate the impact of CB2R activation on protecting hippocampal neurons against Aβ-induced neuronal hyperexcitation.
CB2R is one of the main components of the endogenous cannabinoid system (3). CB2R has been considered a “peripheral” cannabinoid receptor. However, relatively low CB2R expression is also observed in different brain regions under certain conditions (4, 9). Emerging lines of evidence demonstrate that CB2Rs exhibit neuroprotective roles during the process of AD pathogenesis. For example, it has been reported that in an AD model, an increased CB2R number is expressed on microglia surrounding senile plaques and this increased expression is correlated with Aβ1–42 levels and plaque deposition (22, 23). In the context of AD neuroinflammation, there is evidence that CB2R agonists exert anti-inflammatory effects and enhance amyloid removal (35). Furthermore, there is evidence that the activation of CB2R decreases the production of amyloid peptides in a mouse model of AD. Considering these features, CB2Rs appear to be an important target for neuroprotection of AD (26).
This study examines the effects of a selective CB2R agonist, JWH133, on the chronic Aβ1–42 treatment-induced neuronal hyperexcitation in rat primary hippocampal cultures. Our results show that pre-treatment with JWH133 and Aβ1–42 significantly attenuates the AP firing frequency, prolonged AP initiation latency, and increased the AP afterhyperpolarization potential (AHP) compared with oAβ treatment alone, suggesting that the JWH133 well protects hippocampal neurons against the oAβ-induced neuronal hyperexcitation. These effects of JWH133 are abolished by a selective CB2R antagonist AM630, which confirms that it is mediated by the CB2Rs. In addition to neuronal protection when JWH133 and oAβ co-treatment (for 7 days) we also find that even the oAβinduced neuronal hyperexcitation has already established (treated with oAβ alone for 5 day), then, JWH133 (treated with oAβ for 2 days) also abolishes the established neuronal hyperexcitation. Accumulating lines of evidence demonstrate that the activation of CB2Rs exert neuronal protections in various neurological diseases including AD, PD, stroke, and epilepsy (5, 16, 36). The mechanisms of the CB2R-mediated neuroprotection are thought to be mainly mediated through the modulations of neuronal inflammations (6, 24, 31). In this study, provide a novel electrophysiological mechanism to interpret the CB2R-mediated neuroprotection, through a reduction neuronal hyperexcitation. It is well known that the neuronal hyperexcitation can induce an excitatory toxicity and neuronal degeneration (15, 32, 34) and the reduction of neuronal hyperexcitation is a critical strategy to prevent and treat neuronal degeneration and death (18, 19). Based on this concept, the decrease of neuronal hyperexcitation may be a novel therapeutic strategy to prevent or treat AD pathogenesis. For example, we previously report that the inhibition of chronic Aβ-induced neuronal hyperexcitation by the inhibition or knockout of neuronal α7-nAChRs protects the Aβ-induced neuronal toxicity (20, 40). The activation of hippocampal CB2Rs also protects psychiatric behaviors by a reduction hippocampal neuronal excitation and network synchronization (14, 29). In clinical practice, memantine, a NMDA antagonist is a NIH-approved drug to treat AD patients (10, 17). It has been reported that endocannabinoid 2-AG protects Aβ toxicity through CB1R rather than CB2R (12), which may be mediated through different signal pathways and mechanisms. Here we provide new experimental evidence that the selective activation of hippocampal CB2Rs plays an important role in reduction of the Aβ-induced neuronal hyperexcitation and toxicity. Therefore, the CB2R agonist may serve as a new candidate for prevention and treatment of AD.
|
|
CONCLUSIONS |
|---|
This study demonstrates that the selective activation of hippocampal CB2Rs not only prevents oAβ-increased neuronal hyperexcitation, but also abolishes the chronic oAβ established neuronal hyperexcitation, which underlies our recent findings that CB2Rs protect hippocampal neurons against the Aβ-induced neuronal toxicity and degeneration. This novel finding suggests a potentially therapeutic strategy for the treatment of AD using CB2R agonists.
|
|
ACKNOWLEDGMENTS |
|---|
This research was funded by the CNSF (81371437), the Key Area Research and Development Program of Guangdong Province (2018B030334001), and the 2020 Li Ka Shing Foundation Cross-Disciplinary Research Grant (2020LKSFG01A).
|
|
CONFLICTS OF INTEREST |
|---|
The authors declare no conflict of interest.
|
|
AUTHOR CONTRIBUTIONS |
|---|
Conceptualization, J.W. and Z.M.; investigation, Y.Z., J.Z., L.S., S.L., M.W., W.L.; writing—original draft preparation, Y.Z.; writing—review and editing, J.W. and Z.M.; All authors have read and agreed to the published version of the manuscript.
|
|
DATA AVAILABILITY STATEMENT |
|---|
All data generated or analyzed during this study are included in this published article. The datasets used and or analyzed during the current study are available from the corresponding author on reasonable request.
|
|
REFERENCES |
|---|