| Review Article |
|
|
1Department of Cardiovascular Medicine and Regeneration, Matsumoto, Japan;
2Division of Cardiovascular Sciences, Department of Organ Regeneration, Shinshu University Graduate School of Medicine, Matsumoto, Japan
Corresponding author:Masafumi Takahashi, MD, PhD, Division of Cardiovascular Sciences, Department of Organ Regeneration, Shinshu University Graduate School of Medicine, 3-1-1 Asahi, Matsumoto, Nagano 390-8621, Japan. Tel: +81-263-37-3352; Fax: +81-263-37-2573; E-mail: masafumi@sch.md.shinshu-u.ac.jp.
| |
ABSTRACT |
|
|
INTRODUCTION |
|
|
CHEMOKINES |
|
|
THERAPEUTIC IMPLICATIONS |
|
|
CONCLUSION |
|
|
ACKNOWLEDGEMENTS |
|
|
CONFLICT OF INTEREST |
|
|
REFERENCES |
|
|
ABSTRACT
|
|---|
Myocardial infarction (MI) is accompanied by inflammatory responses that lead to the recruitment of leukocytes and subsequent myocardial damage, healing, and scar formation. Chemokines are a family of potent chemoattractant cytokines that regulate the leukocyte trafficking in basal levels and inflammatory processes; however, it has been recently recognized that chemokines are expressed by non-hematopoietic cells such as endothelial cells, smooth muscle cells, and cardiomyocytes, and their function extends far beyond leukocyte migration and activation. Many experimental and clinical studies have demonstrated that chemokines play an important role in the pathophysiology of MI. In particular, the CC chemokine – monocyte chemoattractant protein-1 (MCP-1/CCL2) – is one of the most frequently investigated, and it is believed to play an important role in the pathophysiology of MI. This review will focus on the role of MCP-1 in the pathophysiology of MI and discuss its potential as a therapeutic target in this condition.
KEY WORDS:
cytokines; inflammation; ischemia; leukocyte; myocardium; reperfusion
|
|
INTRODUCTION |
|---|
Myocardial infarction (MI) is a common occurrence and it is predicted that this will be the leading cause of death worldwide in the near future. MI is defined as the necrosis of cardiovascular tissue due to ischemia that results in the replacement of the myocardium by a dense fibrotic scar. The major functional consequence of MI is a decrease in systolic compliance, i.e., progressive loss of pump function in the chamber where the loss of muscle had occurred. In addition, MI often results in electrical instability within the heart and generation of fatal arrhythmias. Therefore, the development of MI may be an important issue that needs to be studied in the 21st century.
The most common cause of MI is atherosclerosis in the coronary arteries (1). The atherosclerotic plaques may become unstable, may rupture, and facilitate the formation of a thrombus that occludes the coronary artery; this leads to MI. In humans, myocardial necrosis begins in the subendocardium at 30–¬¬40 minutes after the onset of coronary occlusion: an almost complete infarction of the area at risk develops after approximately 4 hours of coronary occlusion. However, the exact time for complete infarction is influenced by several other factors including collateral circulation, prior ischemic events, and neurologic reflexes. Furthermore, increased fibrinolytic activity may lead to restoration of the coronary blood flow. In such cases, coronary blood flow is restored and causes “reperfusion injury.” On MI occurrence, the infarcted myocardium is injured by the ischemic insult and this result in myocardial cell death, i.e., necrosis and apoptosis. Simultaneously, circulating leukocytes are recruited to the site of the infarcted area, and subsequently, healing and scar formation occurs. The ischemic insult begins immediately after the coronary occlusion that lasts for several hours: subsequent leukocyte recruitment is also observed. Myocardial healing and myocardial scar formation begins at approximately 72 hours and 1 week, respectively, after the onset of the ischemic insult. In addition, concomitant angiogenesis might influence the degree of myocardial damage and remodeling after MI.
Recent evidence indicates that MI is accompanied by inflammatory responses that lead to the recruitment of leukocytes and subsequent myocardial damage, healing, and scar formation (2). Furthermore, the leukocytes recruited to the site of the infarcted myocardium lead to the release of cytokines and proteinases that may induce further inflammation and left ventricular remodeling. These cells also secrete a large amount of angiogenic factors such as the vascular endothelial growth factor (VEGF) that can induce angiogenesis in the infarcted heart. Thus, inflammation plays a role in the pathological processes from the initiation to the end of MI. These processes include: myocardial necrosis and apoptosis, leukocyte recruitment, myocardial healing and scar formation, and angiogenesis.
Chemokines are a family of potent chemoattractant cytokines that regulate leukocyte trafficking in basal levels and inflammatory processes (3, 4). Depending on its topical concentration, they directly recruit circulating leukocytes to the site of inflammation or injury. Many experimental and clinical studies have demonstrated that a substantial number of chemokines are involved in the pathophysiology of MI. Among these, monocyte chemoattractant protein-1 (MCP-1, also known as CCL2) is one of the most frequently investigated, and it is believed to play an important role in the process of MI. Indeed, during a 10-months follow-up in a large study involving patients with acute coronary syndromes, elevated serum MCP-1 levels were shown to be associated with an increased risk of death or MI (5). Furthermore, a recent study demonstrated that MCP-1 gene polymorphisms are associated with increased serum MCP-1 levels and prevalent MI in the Framingham Heart Study (6). Since MCP-1 mainly recruits monocytes to the site of inflammation and the infarcted area (7), it may play a role in the early stages of atherosclerosis or in the subacute phase of MI. Recently, other investigators and we demonstrated that MCP-1 plays a significant role in the process of myocardial healing and remodeling after MI occurrence (8-11). This review will focus on the role of MCP-1 in the pathophysiology of MI and discuss the potential of MCP-1 as a therapeutic target in MI.
|
|
CHEMOKINES
|
|---|
It has been demonstrated that a number of factors and molecules are involved in the pathophysiology of MI. Among these, chemokines are thought to be one of the key regulators because they have not only been shown to be expressed in the infarcted myocardium but they also play a crucial role in the process of myocardial inflammation and healing (12, 13). In humans, more than 50 chemokines have been identified and divided into two main subfamilies on the basis of the conserved structural features. In the CXC chemokines, a single amino acid separates the two amino¬–terminal cysteine residues; however, in the CC chemokines, no amino acid separates the two cysteines. Other minor chemokine subfamilies that are currently known include fractalkine/CX3CL1 (CX3C chemokines) and lymphotactin/XCL1. Generally, CC chemokines are potent chemoattractants and activators of monocytes and lymphocytes, whereas CXC chemokines attract neutrophils.
Chemokines mediate their effects via interaction with specific chemokines receptors that are expressed on a wide range of cell types. The chemokine receptors are seven transmembrane G-protein-coupled receptors, and are unusual among the many characterized members of the seven transmembrane receptor superfamily in that a single receptor possesses multiple high-affinity ligands for it. Therefore, initially, the chemokine network might appear like a redundant cellular signaling system; however, analysis of chemokine or its receptor gene disruption in mice has revealed that certain chemokines possess unique and non-redundant roles in leukocyte trafficking, inflammation, and immunity.
The basic role of chemokines involves regulation of leukocyte transport and trafficking in basal and inflammatory processes; however, recently, it has been observed that chemokines are also expressed by non-hematopoietic cells such as endothelial cells, smooth muscle cells, and cardiomyocytes, and their function extends far beyond leukocyte migration and activation. For example, the CXC chemokines exert angiogenic or angiostatic effects on endothelial cells. Strieter et al. (14) reported that CXC chemokines containing the ELR (glutamate-leucine-arginine) motif such as interleukin-8 (IL-8, known as CXCL8) stimulate the migration of endothelial cells and promote angiogenesis. In contrast, other CXC chemokines such as platelet factor 4 (PF-4, known as CXCL4) and interferon-inducible protein 10 (IP-10, known as CXCL10) fail to induce endothelial migration or angiogenesis but exert angiostatic effects in the presence of other angiogenic factors (14). Thus, chemokines are key mediators not only in inflammatory responses but also in other responses in the pathophysiology of diseases.
Chemokines involved in myocardial infarction
Many experimental and clinical studies have shown that chemokines are involved in the pathophysiology of MI. The CXC chemokines IL-8 (15-18), stromal cell-derived factor-1 (SDF-1, known as CXCL12) (19-21), GRO-a/KC (known as CXCL1) (16), and IP-10, and the CC chemokines MCP-1 (7, 9, 15, 22-25) and macrophage inflammatory protein-1a/b (MIP-1a/b, known as CCL3/4) (23, 26) appear to be consistently up-regulated in various animal models of experimental MI (Table 1). Among these, two types of MI models are currently used, i.e., permanent MI and ischemia-reperfusion injury models. Actually, several differences exist between the pathophysiology of permanent MI and ischemia-reperfusion injury (27, 28). Reperfusion releases a large excess of oxygen-derived free radicals and causes “reperfusion injury.” Inflammatory responses such as the infiltration of neutrophils and macrophages are much stronger in the reperfused heart than in the infarcted heart. Since the inflammatory responses after MI determine for tissue healing (2), collagen deposition during MI may be accelerated in the reperfused heart. Furthermore, reperfusion enhances neovascularization to a greater extent in the reperfused heart than in the infracted heart. Therefore, it is necessary to pay attention to the MI models used in the studies. Consistent with the results of the experimental studies, elevated levels of MCP-1 were detected in the sera obtained from MI patients (Table 2) (29-34); this indicates the clinical significance of these chemokines in the pathophysiology of MI.
Since CXC chemokines predominantly recruit neutrophils to the infarcted myocardium, they may play a role in myocardial injury after ischemia-reperfusion. With regard to IL-8, recombinant IL-8 markedly increased the adhesion of neutrophils to isolated cardiomyocytes, resulting in direct cytotoxicity for the cells (35); this suggests a potential role in neutrophil-mediated myocardial injury. Furthermore, neutralization of IL-8 significantly reduces the degree of necrosis in a rabbit model of myocardial ischemia-reperfusion injury (36). Therefore, IL-8 is assumed to be deleterious for MI although it has angiogenic effects. Another reported CXC chemokine SDF-1 and its receptor CXCR4 play a critical role in cardiovascular development and angiogenesis (37, 38). We recently found that SDF-1 is markedly upregulated in the infarcted heart, and that CXCR4 cells mobilized by the macrophage colony-stimulating factor (M-CSF) are recruited to the infarcted myocardium, resulting in improved cardiac dysfunction and remodeling after MI (39). In support of this evidence, very recently, Misao et al. (40) reported that the SDF-1–CXCR4 axis contributes to the beneficial effects of granulocyte colony-stimulating factor (G-CSF) in a rabbit model of myocardial ischemia-reperfusion injury. The expression of GRO-a/KC, lipopolysaccharide-induced CXC chemokine (LIX, known as CXCL5), and IP-10 was shown to have increased after MI (41, 42); however, the precise role of these CXC chemokines in MI remains unclear. On the other hand, since CC chemokines mainly recruit monocytes and lymphocytes to the infracted myocardium, they may play a role in the process of healing and scar formation after MI. MIP-1a/b is also upregulated in the infarcted myocardium (26); however, its role remains to be elucidated.
The mechanisms responsible for chemokine upregulation in the infarcted heart have not been fully understood; however, the factors implicated in initiating the inflammatory responses are likely to stimulate the induction of these chemokines. IL-8 and MCP-1 induction is regulated by a transcription factor NF-kB, which contributes to the regulation of inflammation. Recently, it was shown in rats and mice that NF-kB is activated in the infarcted heart and that its inactivation improves cardiac dysfunction and remodeling after MI (43, 44); this suggests the involvement of NF-kB in the pathophysiology of MI. Since inflammatory cytokines such as tumor necrosis factor-a (TNF-a) and interleukin-1b (IL-1b), and reactive oxygen species (ROS) activate NF-kB in cardiomyocytes, these factors may contribute to NF-kB activation in the infarcted myocardium. SDF-1 induction is regulated by another transcription factor, i.e., hypoxia-inducible factor-1a (HIF-1a), which was induced by hypoxia (45). HIF-1 was also reported to be in the active form in the infarcted myocardium after MI (46).
MCP-1 in myocardial infarction
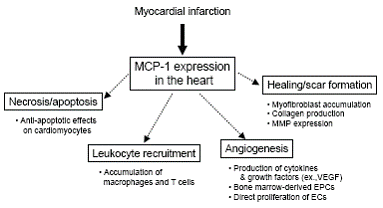
MCP-1 is one of the best-studied CC chemokines that is involved in pathophysiology of MI. MCP-1 induces the recruitment and activation of monocytes, T cells, and NK cells but not neutrophils; it has been implicated in diseases characterized by monocyte-rich infiltrates. MCP-1 is a major ligand for the CC chemokine receptor 2 (CCR2) and binds only to CCR2. CCR2 is one of CC chemokine receptors that are G-protein-coupled receptors with seven transmembrane domains (47). Activation of CCR2 by MCP-1 induces its dimerization, internalization, and activation of downstream signaling pathways including G-proteins, JAK/STAT, mitogen-activated protein (MAP) kinase pathways (48-50); however, little is known about the mechanism of MCP-1 signal transduction via CCR2 in the myocardium. MCP-1 is produced by a variety of cells in response to injury or exposure to other cytokines, such as IL-1b, IL-6, and TNF-a, and has been implicated in heart failure, ischemia-reperfusion, and MI (13, 51). Several transcription factors have been proposed for the regulation of MCP-1. In human MCP-1 gene, tissue-type plasminogen activator (t-PA)-responsive elements (TRE) and kB enhancer element exist at the 5’-flanking region of MCP-1 gene; this suggest that the role of activator protein-1 (AP-1) and NF-kB (52). Further, several signaling molecules are involved in the activation of MCP-1 gene, including protein kinase C and tyrosine kinases (53). Recent evidence suggests that MCP-1 may affect many processes involved in MI, namely, myocardial necrosis and apoptosis, leukocyte recruitment, myocardial healing and scar formation, and angiogenesis.
Myocardial necrosis and apoptosis
In acute MI, a large number of cardiomyocytes die as a result of apoptosis and necrosis (54-56). Recent studies suggest that ischemia-reperfusion, not ischemia alone, may be required to complete the apoptotic process in cardiomyocytes. Zhao et al. (56) discovered that prolonged ischemia without reperfusion induces necrosis but not apoptosis, whereas ischemia followed by reperfusion induces apoptosis. Another report confirmed that the apoptotic process is triggered by ischemia and reperfusion is required to complete this process (55).
Since monocyte/macrophage recruitment to the infarcted myocardium was observed in the subacute phase of MI, it is unlikely that the monocyte chemoattractant effects of MCP-1 contribute to this process. However, recent investigations suggest that MCP-1 affects other types of cells such as cardiomyocytes and endothelial cells (57-60). In particular, Tarzami et al. (60, 61) recently tested the effect of MCP-1 on apoptosis induced by hypoxia in neonatal rat cultured cardiomyocytes and reported that MCP-1 protects the cardiomyocytes from hypoxia-induced apoptosis. The anti-apoptotic effect of MCP-1 is mediated via several pathways: extracellular-regulated kinase 1/2 (ERK1/2) and Bcl-2 family proteins, but not Gai. Interestingly, the monocyte chemoattractant effect is mediated by the activation of the G protein Gai (62); this process is inhibited by the pertussis toxin. Therefore, anti-apoptotic and monocyte chemoattractant effects of MCP-1 may be mediated via the different signaling pathways. The in vivo role of MCP-1 in apoptosis and/or necrosis in the infarcted heart is currently unclear.
Leukocyte recruitment
Since MCP-1 is a potent chemoattractant for monocytes, MCP-1 plays a substantial role in the recruitment of monocytes/macrophages to the infarcted myocardium. Since the chemoattractant effect of MCP-1 is regulated by its topical concentrations (63, 64), we investigated the role of cardiac MCP-1 that is topically produced in the infarcted heart; transgenic mice expressing the mouse JE-MCP-1 gene under the control of the a-cardiac myosin heavy chain promoter (MHC/MCP-1 mice) were used for this purpose (8). At 12 weeks, the MHC/MCP-1 mice showed a slight increased in macrophage infiltration in the heart; however, no cardiac dysfunction and hypertrophy were observed. After MI, the macrophage infiltration increased in the infarcted area of the heart in wild-type mice and it markedly increased in MHC/MCP-1 mice. Concomitantly, capillary formation is also promoted in the infarcted heart of MHC/MCP-1 mice. Hayashidani et al. (9) reported that anti-MCP-1 gene therapy markedly reduced the infiltration of macrophages but not that of T cells in the infarcted myocardium. Dewald et al. (10) also reported that the infiltration of macrophages, but not neutrophils, was suppressed and delayed in MCP-1-null mice when compared with that in wild-type mice. These findings indicate that MCP-1 is a critical factor for the recruitment of monocytes/macrophages in the infarcted heart.
Myocardial healing and scar formation
Macrophages and myofibroblasts play a crucial role in the process of myocardial repair after MI. Macrophages phagocytose the necrotic myocardium and concomitantly, myofibroblasts proliferate and migrate into the infarcted area (2, 65). The necrotic tissue is replaced by the granulation tissue. As repair proceeds, myofibroblasts secrete and deposit collagen and other extracellular matrixes; subsequently, apoptosis of the granulation tissue cells results in the formation of a thin and hypocellular scar formation (66). Progressive cardiac wall thinning and chamber dilatation (remodeling) are associated with the increased incidence of congestive heart failure, aneurysm formation, and mortality.
MCP-1 may influence the replacement of granulation tissue and myofibroblast accumulation in the healing process after MI. MCP-1 directly modulates fibroblast activity by increasing collagen (67) and matrix metalloproteinase expression (68). Dewald et al. (10) reported that in the infarcted heart of murine ischemia-reperfusion models, MCP-1 deficiency delays macrophage recruitment and replacement of cardiomyocytes by the granulation tissue. They further demonstrated that after MI, MCP-1 deficiency diminished myofibroblast accumulation and attenuated left ventricular remodeling. We recently showed that MCP-1 directly promotes the in vitro differentiation of cardiac fibroblasts into myofibroblasts (8). In addition, the hypoxic conditions increased the differentiation of cardiac fibroblasts and it was further enhanced in the presence of MCP-1. In fact, after MI, myofibroblast accumulation had notably increased in the infarcted myocardium in MHC/MCP-1 mice in contrast to that observed in wild-type mice. Taken together, these findings suggest a critical role for MCP-1 in myocardial healing, scar formation, and remodeling after MI.
Angiogenesis
Angiogenesis could affect infarct size and myocardial remodeling by supplying oxygen and nutrients necessary to maintain metabolism. Recent evidence indicates that MCP-1 promotes the in vitro and in vivo formation of new blood vessels in ischemic tissues. Weber et al. (58) reported that human endothelial cells express CCR2, which is upregulated by inflammatory cytokines, and MCP-1 induces migration and proliferation of the endothelial cells. After mechanical injury to the endothelial monolayers, which spontaneously closed within 24 hours, wound repair was delayed by a CCR2 antagonist and an MCP-1 blocking antibody. Similarly, using the chick chorioallantoic membrane (CAM) and matrigel plug assays, Salcedo et al. (57) showed that MCP-1 induces in vivo formation of new blood vessels. In addition, Schwarz et al. (69) tested the effects of MCP-1 on angiogenesis (newly developed capillaries) and arteriogenesis (the development of functionally active arterioles from pre-existing blood vessels) in a chronic MI rat model; they showed that an intramyocardial injection of recombinant MCP-1 into the infarct border zone induces monocyte infiltration and angiogenesis but not arteriogenesis in the infarcted heart. Recently, we also observed in MHC/MCP-1 mice that capillary formation and macrophage infiltration significantly increased after MI. Moreover, Low et al. (70) demonstrated that MCP-1–/– mice exhibit delayed wound angiogenesis demonstrating lower capillary density than their wild-type littermates; this suggests an important role of this chemokine in wound healing and concurrent angiogenesis.
Several possible mechanisms have been postulated for the induction of angiogenesis by MCP-1. First, MCP-1 directly upregulates HIF-1a gene expression and subsequently stimulates VEGF induction by endothelial cells (71). MCP-1 has also been shown to stimulate VEGF induction by macrophage recruitment to the ischemic tissues (72). Second, recent evidence indicates that the bone marrow-derived monocytic lineage cells function as endothelial progenitor cells (EPCs) and participate in the formation of new blood vessels in the ischemic tissues. Our recent study showed that in contrast to wild-type mice, MHC/MCP-1 mice did not show an increase in the EPCs (CD34+/Flk-1+ cells; known as an ordinary EPC marker (73)) in the peripheral circulation after MI; this suggests that the monocytic cell-derived EPCs may have other surface markers. In this regard, Harraz et al. (74) reported that the CD34-negative angioblasts are a subset of the CD14-positive monocytic cells, and these monocytes have the potential to transdifferentiate into endothelial cells. Fujiyama et al. (75) reported that the bone marrow-derived CD34–/CD14+ EPCs adhere to the injured endothelium of a rat carotid artery in an MCP-1-dependent manner and accelerate endothelial repair; this suggests that MCP-1 may stimulate bone marrow-derived monocytic EPCs. Third, interesting findings by Moldovan and his colleagues (76) revealed that the monocytes/macrophages accumulated by MCP-1 produce matrix metalloproteinase-dependent tunnels and promote the formation of new vessels. In addition, more recently, they demonstrated that monocytes/macrophages are required to induce efficient angiogenesis and tissue repair besides inducing parent endothelial cells or EPCs (77). Taken together, MCP-1 regulates the process of angiogenesis in the infarcted myocardium, thereby influencing cardiac function and remodeling after MI.
|
|
THERAPEUTIC IMPLICATIONS
|
|---|
The Framingham Heart Study demonstrated that variations in the MCP-1 gene influence serum MCP-1 levels and the incidence of myocardial infarction (6). Other clinical studies have also indicated that higher serum MCP-1 levels are associated with an increased risk of myocardial infarction, sudden cardiac death, coronary angioplasty, and stent restenosis (5, 78, 79). Thus, MCP-1 may be a potential target for therapeutic interventions in MI. Indeed, several experimental studies have suggested that the inhibition of MCP-1 signaling improves cardiac dysfunction and remodeling after MI (9, 11), suggesting a deleterious effect of MCP-1 in MI. In contrast, other investigators and we demonstrated that cardiac overexpression of MCP-1 improves cardiac dysfunction and remodeling after MI (8, 80). Furthermore, recent studies have shown that the role of MCP-1 extends beyond its monocyte chemoattractant effects; it also induces angiogenic and cardioprotective effects (57, 60, 61, 71). Since MCP-1 is produced in the infarcted heart (7, 81) and the effects of MCP-1 are regulated by its topical concentration (63, 64), the beneficial or deleterious effects of MCP-1 might depend on the situation, i.e. local concentration, duration, and time period after MI.
|
|
|
|
|
CONCLUSIONS
|
|---|
A growing number of studies have indicated that chemokines are key mediators of MI. Of these, MCP-1 is one of the most important chemokines that is involved in the pathophysiology of MI. In this review, we discussed the role of MCP-1 in the processes of MI; myocardial necrosis and apoptosis, leukocyte recruitment, myocardial healing and scar formation, and angiogenesis. Further, MCP-1 is thought to be a potential target for the treatment of MI; however, prior to its clinical application of MCP-1, further investigations are necessary to elucidate its precise role in MI.
|
|
ACKNOWLEDGEMENTS
|
|---|
This study was supported by research grants from the Ministry of Health, Labor and Welfare of Japan (Research on Measures for Intractable Diseases) and the Ministry of Education, Science, Sports and Culture.
|
|
CONFLICT OF INTEREST
|
|---|
The authors declare that no conflicting interests exist.
|
|
REFERENCES
|
|---|
1. Fallon JT. Pathology of myocardial infarction and reperfusion. Philadelphia: Lippincott-Raven Publishers, 1996.
2. Frangogiannis NG, Smith CW, Entman ML. The inflammatory response in myocardial infarction. Cardiovasc Res 2002; 53:31-47.
3. Gerard C, Rollins BJ. Chemokines and disease. Nat Immunol 2001; 2:108-15.
4. Baggiolini M. Chemokines and leukocyte traffic. Nature 1998; 392:565-568.
5. de Lemos JA, Morrow DA, Sabatine MS, Murphy SA, et al. Association between plasma levels of monocyte chemoattractant protein-1 and long-term clinical outcomes in patients with acute coronary syndromes. Circulation 2003; 107:690-695.
6. McDermott DH, Yang Q, Kathiresan S, Cupples LA, et al. CCL2 polymorphisms are associated with serum monocyte chemoattractant protein-1 levels and myocardial infarction in the Framingham Heart Study. Circulation 2005; 112:1113-1120.
7. Kumar AG, Ballantyne CM, Michael LH, Kukielka GL, et al. Induction of monocyte chemoattractant protein-1 in the small veins of the ischemic and reperfused canine myocardium. Circulation 1997; 95:693-700.
8. Morimoto H, Takahashi M, Izawa A, Ise H, et al. Cardiac overexpression of monocyte chemoattractant protein-1 in transgenic mice prevents cardiac dysfunction and remodeling after myocardial infarction. Circ Res 2006; 99:891-899.
9. Hayashidani S, Tsutsui H, Shiomi T, Ikeuchi M, et al. Anti-monocyte chemoattractant protein-1 gene therapy attenuates left ventricular remodeling and failure after experimental myocardial infarction. Circulation 2003; 108:2134-2140.
10. Dewald O, Zymek P, Winkelmann K, Koerting A, et al. CCL2/Monocyte Chemoattractant Protein-1 regulates inflammatory responses critical to healing myocardial infarcts. Circ Res 2005; 96:881-889.
11. Kaikita K, Hayasaki T, Okuma T, Kuziel WA, et al. Targeted deletion of CC chemokine receptor 2 attenuates left ventricular remodeling after experimental myocardial infarction. Am J Pathol 2004; 165:439- 447.
12. Frangogiannis NG. Chemokines in the ischemic myocardium: from inflammation to fibrosis. Inflamm Res 2004; 53:585-595.
13. Frangogiannis NG, Entman ML. Chemokines in myocardial ischemia. Trends Cardiovasc Med 2005; 15:163-169.
14. Strieter RM, Polverini PJ, Kunkel SL, Arenberg DA, et al. The functional role of the ELR motif in CXC chemokine-mediated angiogenesis. J Biol Chem 1995; 270:27348-27357.
15. Tonnessen T, Florholmen G, Henriksen UL, Christensen G. Cardiopulmonary alterations in mRNA expression for interleukin-1beta, the interleukin-6 superfamily and CXC-chemokines during development of postischaemic heart failure in the rat. Clin Physiol Funct Imaging 2003; 23:263-268.
16. Kocher AA, Schuster MD, Bonaros N, Lietz K, et al. Myocardial homing and neovascularization by human bone marrow angioblasts is regulated by IL-8/Gro CXC chemokines. J Mol Cell Cardiol 2006; 40:455-464.
17. Ivey CL, Williams FM, Collins PD, Jose PJ, et al. Neutrophil chemoattractants generated in two phases during reperfusion of ischemic myocardium in the rabbit. Evidence for a role for C5a and interleukin-8. J Clin Invest 1995; 95:2720-2728.
18. Kilgore KS, Park JL, Tanhehco EJ, Booth EA, et al. Attenuation of interleukin-8 expression in C6-deficient rabbits after myocardial ischemia/ reperfusion. J Mol Cell Cardiol 1998; 30:75-85.
19. Abbott JD, Huang Y, Liu D, Hickey R, et al. Stromal cell-derived factor- 1alpha plays a critical role in stem cell recruitment to the heart after myocardial infarction but is not sufficient to induce homing in the absence of injury. Circulation 2004; 110:3300-3305.
20. Ma J, Ge J, Zhang S, Sun A, et al. Time course of myocardial stromal cell-derived factor 1 expression and beneficial effects of intravenously administered bone marrow stem cells in rats with experimental myocardial infarction. Basic Res Cardiol 2005; 100:217-223.
21. Pillarisetti K, Gupta SK. Cloning and relative expression analysis of rat stromal cell derived factor-1 (SDF-1)1: SDF-1 alpha mRNA is selectively induced in rat model of myocardial infarction. Inflammation 2001; 25:293-300.
22. Wayman NS, Hattori Y, McDonald MC, Mota-Filipe H, et al. Ligands MC P-1 in myocardial infarction of the peroxisome proliferator-activated receptors (PPAR-gamma and PPAR-
23. Maekawa N, Wada H, Kanda T, Niwa T, et al. Improved myocardial ischemia/reperfusion injury in mice lacking tumor necrosis factoralpha. J Am Coll Cardiol 2002; 39:1229-1235.
24. Lu L, Zhang JQ, Ramires FJ, Sun Y. Molecular and cellular events at the site of myocardial infarction: from the perspective of rebuilding myocardial tissue. Biochem Biophys Res Commun 2004; 320:907- 913.
25. Lakshminarayanan V, Lewallen M, Frangogiannis NG, Evans AJ, et al. Reactive oxygen intermediates induce monocyte chemotactic protein- 1 in vascular endothelium after brief ischemia. Am J Pathol 2001; 159:1301-1311.
26. Nossuli TO, Frangogiannis NG, Knuefermann P, Lakshminarayanan V, et al. Brief murine myocardial I/R induces chemokines in a TNFalpha- independent manner: role of oxygen radicals. Am J Physiol Heart Circ Physiol 2001; 281:H2549-2558.
27. Vandervelde S, van Amerongen MJ, Tio RA, Petersen AH, et al. Increased inflammatory response and neovascularization in reperfused vs. non-reperfused murine myocardial infarction. Cardiovasc Pathol 2006; 15:83-90.
28. Kloner RA, Jennings RB. Consequences of brief ischemia: stunning, preconditioning, and their clinical implications: part 1. Circulation 2001; 104:2981-2989.
29. Soejima H, Ogawa H, Yasue H, Kaikita K, et al. Angiotensin-converting enzyme inhibition reduces monocyte chemoattractant protein-1 and tissue factor levels in patients with myocardial infarction. J Am Coll Cardiol 1999; 34:983-988.
30. Murakami Y, Kurosaki K, Matsui K, Shimada K, et al. Serum MCP-1 and VEGF levels are not affected by inhibition of the renin-angiotensin system in patients with acute myocardial infarction. Cardiovasc Drugs Ther 2003; 17:249-255.
31. Wang G, Mao JM, Wang X, Zhang FC. Effect of homocysteine on plaque formation and oxidative stress in patients with acute coronary syndromes. Chin Med J (Engl) 2004; 117:1650-1654.
32. Kameda K, Matsunaga T, Abe N, Fujiwara T, et al. Increased pericardial fluid level of matrix metalloproteinase-9 activity in patients with acute myocardial infarction: possible role in the development of cardiac rupture. Circ J 2006; 70:673-678.
33. Iwata A, Miura S, Shirai K, Kawamura A, et al. Lower level of lowdensity lipoprotein cholesterol by statin prevents progression of coronary restenosis after successful stenting in acute myocardial infarction. Intern Med 2006; 45:885-890.
34. Arakelyan A, Petrkova J, Hermanova Z, Boyajyan A, et al. Serum levels of the MCP-1 chemokine in patients with ischemic stroke and myocardial infarction. Mediators Inflamm 2005; 2005:175-179.
35. Kukielka GL, Smith CW, LaRosa GJ, Manning AM, et al. Interleukin- 8 gene induction in the myocardium after ischemia and reperfusion in vivo. J Clin Invest 1995; 95:89-103.
36. Boyle EM, Jr., Kovacich JC, Hebert CA, Canty TG, Jr., et al. Inhibition of interleukin-8 blocks myocardial ischemia-reperfusion injury. J Thorac Cardiovasc Surg 1998; 116:114-121.
37. Nagasawa T, Hirota S, Tachibana K, Takakura N, et al. Defects of Bcell lymphopoiesis and bone-marrow myelopoiesis in mice lacking the CXC chemokine PBSF/SDF-1. Nature 1996; 382:635-638.
38. Salvucci O, Yao L, Villalba S, Sajewicz A, et al. Regulation of endothelial cell branching morphogenesis by endogenous chemokine stromal- derived factor-1. Blood 2002; 99:2703-2711.
39. Morimoto H, Takahashi M, Shiba Y, Ise H, et al. Bone marrow-derived CXCR4-positive cells mobilized by macrophage colony-stimulating factor participate in the prevention of cardiac dysfunction after myocardial infarction in mice. Circulation 2006; 114:II-171.
40. Misao Y, Takemura G, Arai M, Ohno T, et al. Importance of recruitment of bone marrow-derived CXCR4+ cells in post-infarct cardiac repair mediated by G-CSF. Cardiovasc Res 2006; 71:455-465.
41. Chandrasekar B, Smith JB, Freeman GL. Ischemia-reperfusion of rat myocardium activates nuclear factor-KappaB and induces neutrophil infiltration via lipopolysaccharide-induced CXC chemokine. Circulation 2001; 103:2296-2302.
42. Frangogiannis NG, Mendoza LH, Lewallen M, Michael LH, et al. Induction and suppression of interferon-inducible protein 10 in reperfused myocardial infarcts may regulate angiogenesis. FASEB J. 2001; 15:1428-1430.
43. Kawano S, Kubota T, Monden Y, Tsutsumi T, et al. Blockade of NFkappaB improves cardiac function and survival after myocardial infarction. Am J Physiol Heart Circ Physiol 2006; 291:H1337-1344.
44. Onai Y, Suzuki J, Maejima Y, Haraguchi G, et al. Inhibition of NF- {kappa}B improves left ventricular remodeling and cardiac dysfunction after myocardial infarction. Am J Physiol Heart Circ Physiol. 2006.
45. Ceradini DJ, Kulkarni AR, Callaghan MJ, Tepper OM, et al. Progenitor cell trafficking is regulated by hypoxic gradients through HIF-1 induction of SDF-1. Nat Med 2004; 10:858-864.
46. Ceradini DJ, Gurtner GC. Homing to hypoxia: HIF-1 as a mediator of progenitor cell recruitment to injured tissue. Trends Cardiovasc Med 2005; 15:57-63.
47. Charo IF, Myers SJ, Herman A, Franci C, et al. Molecular cloning and functional expression of two monocyte chemoattractant protein 1 receptors reveals alternative splicing of the carboxyl-terminal tails. Proc Natl Acad Sci U S A 1994; 91:2752-2756.
48. Arai H, Charo IF. Differential regulation of G-protein-mediated signaling by chemokine receptors. J Biol Chem 1996; 271:21814-21819.
49. Mellado M, Rodriguez-Frade JM, Vila-Coro AJ, Fernandez S, et al. Chemokine receptor homo- or heterodimerization activates distinct signaling pathways. EMBO J. 2001; 20:2497-2507.
50. Rodriguez-Frade JM, Vila-Coro AJ, de Ana AM, Albar JP, et al. The chemokine monocyte chemoattractant protein-1 induces functional responses through dimerization of its receptor CCR2. Proc Natl Acad Sci U S A 1999; 96:3628-3633.
51. Dewald O, Frangogiannis NG, Zoerlein M, Duerr GD, et al. Development of murine ischemic cardiomyopathy is associated with a transient inflammatory reaction and depends on reactive oxygen species. Proc Natl Acad Sci U S A 2003; 100:2700-2705.
52. Shyy YJ, Li YS, Kolattukudy PE. Structure of human monocyte chemotactic protein gene and its regulation by TPA. Biochem Biophys Res Commun 1990; 169:346-351.
53. Shyy YJ, Li YS, Kolattukudy PE. Activation of MCP-1 gene expression is mediated through multiple signaling pathways. Biochem Biophys Res Commun 1993; 192:693-699.
54. Gottlieb RA, Burleson KO, Kloner RA, Babior BM, et al. Reperfusion injury induces apoptosis in rabbit cardiomyocytes. J Clin Invest 1994; 94:1621-1628.
55. Freude B, Masters TN, Robicsek F, Fokin A, et al. Apoptosis is initiated by myocardial ischemia and executed during reperfusion. J Mol Cell Cardiol 2000; 32:197-208.
56. Zhao ZQ, Nakamura M, Wang NP, Wilcox JN, et al. Reperfusion induces myocardial apoptotic cell death. Cardiovasc Res 2000; 45:651- 660.
57. Salcedo R, Ponce ML, Young HA, Wasserman K, et al. Human endothelial cells express CCR2 and respond to MCP-1: direct role of MCP-1 in angiogenesis and tumor progression. Blood 2000; 96:34-40.
58. Weber KS, Nelson PJ, Grone HJ, Weber C. Expression of CCR2 by endothelial cells : implications for MCP-1 mediated wound injury MC P-1 in myocardial infarction repair and In vivo inflammatory activation of endothelium. Arterioscler Thromb Vasc Biol 1999; 19:2085-2093.
59. Ban K, Ikeda U, Takahashi M, Kanbe T, et al. Expression of intercellular adhesion molecule-1 on rat cardiac myocytes by monocyte chemoattractant protein-1. Cardiovasc Res 1994; 28:1258-1262.
60. Tarzami ST, Calderon TM, Deguzman A, Lopez L, et al. MCP-1/ CCL2 protects cardiac myocytes from hypoxia-induced apoptosis by a G(alphai)-independent pathway. Biochem Biophys Res Commun 2005; 335:1008-1016.
61. Tarzami ST, Cheng R, Miao W, Kitsis RN, et al. Chemokine expression in myocardial ischemia: MIP-2 dependent MCP-1 expression protects cardiomyocytes from cell death. J Mol Cell Cardiol 2002; 34:209-221.
62. Cambien B, Pomeranz M, Millet MA, Rossi B, et al. Signal transduction involved in MCP-1-mediated monocytic transendothelial migration. Blood 2001; 97:359-366.
63. Takahashi M, Masuyama J, Ikeda U, Kitagawa S, et al. Suppressive role of endogenous endothelial monocyte chemoattractant protein-1 on monocyte transendothelial migration in vitro. Arterioscler Thromb Vasc Biol 1995; 15:629-636.
64. Takahashi M, Masuyama J, Ikeda U, Kasahara T, et al. Induction of monocyte chemoattractant protein-1 synthesis in human monocytes during transendothelial migration in vitro. Circ Res. 1995; 76:750- 757.
65. Virag JI, Murry CE. Myofibroblast and endothelial cell proliferation during murine myocardial infarct repair. Am J Pathol 2003; 163:2433- 2440.
66. Desmouliere A, Redard M, Darby I, Gabbiani G. Apoptosis mediates the decrease in cellularity during the transition between granulation tissue and scar. Am J Pathol 1995; 146:56-66.
67. Gharaee-Kermani M, Denholm EM, Phan SH. Costimulation of fibroblast collagen and transforming growth factor beta1 gene expression by monocyte chemoattractant protein-1 via specific receptors. J Biol Chem 1996; 271:17779-17784.
68. Yamamoto T, Eckes B, Mauch C, Hartmann K, et al. Monocyte chemoattractant protein-1 enhances gene expression and synthesis of matrix metalloproteinase-1 in human fibroblasts by an autocrine IL-1 alpha loop. J Immunol 2000; 164:6174-6179.
69. Schwarz ER, Meven DA, Sulemanjee NZ, Kersting PH, et al. Monocyte chemoattractant protein 1-induced monocyte infiltration produces angiogenesis but not arteriogenesis in chronically infarcted myocardium. J Cardiovasc Pharmacol Ther 2004; 9:279-289.
70. Low QE, Drugea IA, Duffner LA, Quinn DG, et al. Wound healing in MIP-1alpha(-/-) and MCP-1(-/-) mice. Am J Pathol 2001; 159:457-463.
71. Hong KH, Ryu J, Han KH. Monocyte chemoattractant protein-1- induced angiogenesis is mediated by vascular endothelial growth factor- A. Blood 2005; 105:1405-1407.
72. Niiyama H, Kai H, Yamamoto T, Shimada T, et al. Roles of endogenous monocyte chemoattractant protein-1 in ischemia-induced neovascularization. J Am Coll Cardiol 2004; 44:661-666.
73. Sata M, Nagai R. Inflammation, angiogenesis, and endothelial progenitor cells: how do endothelial progenitor cells find their place? J Mol Cell Cardiol 2004; 36:459-463.
74. Harraz M, Jiao C, Hanlon HD, Hartley RS, et al. CD34- blood-derived human endothelial cell progenitors. Stem Cells 2001; 19:304-312.
75. Fujiyama S, Amano K, Uehira K, Yoshida M, et al. Bone marrow monocyte lineage cells adhere on injured endothelium in a monocyte chemoattractant protein-1-dependent manner and accelerate reendothelialization as endothelial progenitor cells. Circ Res 2003; 93:980- 989.
76. Moldovan NI, Goldschmidt-Clermont PJ, Parker-Thornburg J, Shapiro SD, et al. Contribution of monocytes/macrophages to compensatory neovascularization: the drilling of metalloelastase-positive tunnels in ischemic myocardium. Circ Res 2000; 87:378-384.
77. Anghelina M, Krishnan P, Moldovan L, Moldovan NI. Monocytes/ macrophages cooperate with progenitor cells during neovascularization and tissue repair: conversion of cell columns into fibrovascular bundles. Am J Pathol 2006; 168:529-541.
78. Hojo Y, Ikeda U, Katsuki T, Mizuno O, et al. Chemokine expression in coronary circulation after coronary angioplasty as a prognostic factor for restenosis. Atherosclerosis 2001; 156:165-170.
79. Hojo Y, Ikeda U, Takahashi M, Shimada K. Increased levels of monocyte- related cytokines in patients with unstable angina. Atherosclerosis 2002; 161:403-408.
80. Martire A, Fernandez B, Buehler A, Strohm C, et al. Cardiac overexpression of monocyte chemoattractant protein-1 in transgenic mice mimics ischemic preconditioning through SAPK/JNK1/2 activation. Cardiovasc Res 2003; 57:523-534.
81. Dewald O, Ren G, Duerr GD, Zoerlein M, et al. Of mice and dogs: species-specific differences in the inflammatory response following myocardial infarction. Am J Pathol 2004; 164:665-677.