| Original Article |
|
|
1 Nihon University School of Medicine 2 Division of Receptor Biology, Advanced Medical Research Center 3 Second Department of Internal Medicine Nihon University School of Medicine, Tokyo, Japan
Correspondence to: Tomohiro Nakayama MD, Division of Receptor Biology, Advanced Medical Research Center, Nihon University School of Medicine, Ooyaguchi-kamimachi, 30-1 Itabashi-ku, Tokyo 173-8610, Japan. Tel: +81 3-3972-8111(ext.2751) Fax: +81 3-5375-8076 E-mail: tnakayam@med.nihon-u.ac.jp
Short title: Aminopeptidase A gene and hypertension
| |
ABSTRACT |
| INTRODUCTION | |
|
|
SUBJECTS |
|
|
BIOCHEMICAL ANALYSIS |
|
|
GENOTYPING |
|
|
HAPLOTYPE-BASE CASE CONTROL STUDY |
|
|
STATISTICAL ANALYSIS |
| RESULTS | |
|
|
DISSCUSSION |
|
|
ACKNOWLEDGMENTS |
|
|
REFERENCES |
|
|
ABSTRACT
|
|---|
Aminopeptidase A (APA) cleaves the N-terminal aspartyl acid residue of angiotensin II (Ang II) to produce angiotensin III (Ang III). It has been reported that the APA knockout mouse exhibits elevated blood pressure. Therefore, the APA gene is thought to be a susceptibility gene for essential hypertension (EH). However, extensive studies have yet to define the relationship between the APA gene and EH. The aims of this study were to genotype some of the single nucleotide polymorphisms (SNPs) for the human APA gene and to perform a haplotype-based case-control study to further assess the association between and the APA gene and EH. We performed a genetic association study using SNPs in 227 EH patients and 221 age-matched normotensive (NT) individuals. Although the overall distribution of the genotype did not significantly differ between the EH and NT groups when the entire group of subjects were evaluated, the frequency of rs2290105 did differ between the two when just women were included in the analysis. The haplotype-based case-control analysis also revealed a significant difference between the women of the EH and NT groups. The A-T-A-C haplotype was significantly higher in the EH versus the NT group. These results suggest that rs2290105 and the A-T-A-C haplotype of the APA gene are genetic markers for EH, and that APA or a neighboring gene might be a susceptibility gene for EH.
KEY WORDS: essential hypertension, aminopeptidase A, single nucleotide polymorphism, haplotype, genetic, association study
|
|
INTRODUCTION |
|---|
Angiotensinogen is cleaved by renin to generate angiotensin I (Ang I), which
is further converted to angiotensin II (Ang II) by the angiotensin converting
enzyme (ACE). Ang II is the fundamental peptide of the renin-angiotensin system
(RAS), which induces constriction of blood vessels and increases the sodium and
water retention, thus leading to an increase in blood pressure. Previous studies
using mice that had a disruption of the genes responsible for the generation of
angiotensin and the encoding of Ang II have provided information on the function
of these genes. Aminopeptidase A (APA) cleaves the N-terminal aspartyl acid
residue of Ang II to produce angiotensin III (Ang III). APA is the so-called
angiotensinase and several studies have reported that purified APA functions as
a hypotensive factor and that inhibitors of APA are hypertensive factors in
vivo. A study that examined APA deficient mice found that the baseline values
for systolic blood pressure (SBP) and mean blood pressure (MBP) were higher in
these animals. The blood pressure level for the mice with the targeted
homozygous APA gene (APA-/-) was found to be significantly higher than that seen
in the heterozygous (APA+/-) and wild types (APA+/+). These results suggest that
APA is involved in blood pressure regulation, and that the metabolism of Ang II
is a very important factor in the physiological regulation of blood pressure
[1-5]. Although the APA gene is thought to be a
susceptibility gene for hypertension, there have been no studies examining that
examined the association between the APA gene and essential hypertension (EH).
The APA gene is also listed as the negative primary endpoint (ENPEP) gene in the
NCBI online database. The human APA gene is located on chromosome 4q25. This
gene contains 18 exons, is interrupted by 17 introns, and has several single
nucleotide polymorphisms (SNPs) [6,7]. High
blood pressure or hypertension affects 25% of most adult populations and is an
important risk factor for death from stroke, myocardial infarction and
congestive heart failure. The majority of hypertensive cases are classified as
primary and are referred to as essential hypertension (EH). EH is thought to be
a multifactorial disease [8]. However, there are a few reports
that identify the susceptibility genes of EH as angiotensinogen [9], or
angiotensin converting enzyme [10,11].
The aims of this study were to genotype some of the single nucleotide
polymorphisms (SNPs) for the human APA gene and to perform a haplotype-based
case-control study to further assess the association between and the APA gene
and EH.
Methods
|
|
SUBJECTS
|
|---|
This study included a group of 227 patients that were diagnosed with EH. A positive diagnosis required the patient to have a seated SBP above 160 mmHg and/or diastolic blood pressure (DBP) above 100 mmHg on 3 occasions within 2 months after their first medical examination. None of the patients were using anti-hypertensive medication and any of the subjects diagnosed with secondary hypertension were excluded. We also included 221 normotensive (NT) healthy individuals as controls. None of the NT participants had a family history of hypertension, and they all had SBP and DBP below 130 and 85 mmHg, respectively. A family history of hypertension was defined as prior diagnosis of hypertension in grandparents, uncles, aunts, parents or siblings. Both groups were recruited from the northern area of Tokyo, and informed consent was obtained from each individual as per the protocol approved by the Human Studies Committee of Nihon University[12].
|
|
BIOCHEMICAL ANALYSIS |
|---|
The methodology of the Clinical Laboratory Department of Nihon University Hospital was used to measure all plasma total cholesterol and HDL-cholesterol concentrations, and serum creatinine and uric acid concentrations. [13].
|
|
GENOTYPING |
|---|
Based on the allelic frequency data for the registered SNPs from the National
Center for Biotechnology Information (NCBI) website and from Applied
Biosystems-Celera Discovery System, we chose SNPs with minor allele frequencies
of 20% or greater. This criterion was chosen since SNPs with a high frequency of
the minor allele are very useful as genetic markers in genetic association
studies.
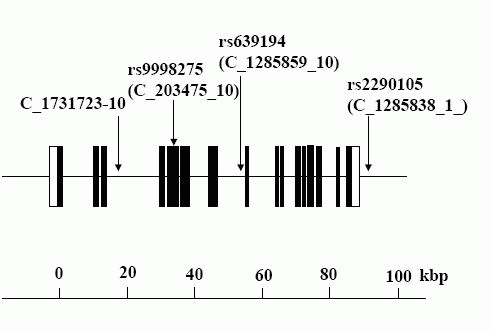
We selected 4 SNPs in the introns of the human APA gene as markers for the
genetic association experiment (Fig. 1). The minor allele frequencies for each
of the SNPs among the Japanese subjects were >10% (screening estimate from the
Celera Company), which indicates that they all should be effective genetic
markers. All SNPs were confirmed using the dbSNP on the NCBI website and the
Applied Biosystems-Celera Discovery System. The accession numbers were as
follows: C_1731723_10 (no registration in the NCBI), rs9998275 (C_203475_10),
rs639194 (C_1285859_10), and rs2290105 (C_1285838_1) (Fig. 1). Genotypes were
determined using Assays-on-Demand kits (Applied Biosystems, Branchburg, NJ)
together with TaqMan○R PCR. When allele-specific fluorogenic probes hybridize to
the template during the polymerase chain reaction (PCR), the 5' nuclease
activity of the Taq polymerase can discriminate alleles. Cleavage results in
increased emission of a reporter dye that otherwise is quenched by the dye
TAMRA. Each 5' nuclease assay requires two unlabeled PCR primers and two
allele-specific probes. Each probe is labeled with a reporter dye (VIC and FAM)
at the 5' end and TAMRA at the 3' end. Amplification by PCR was carried out
using TaqMan Universal Master Mix (PE Biosystems) in a 25 u1 reaction volume
with final total concentrations of 50 ng DNA, 700 nM primer, and 100 nM probe.
Thermal cycling conditions consisted of 95°C for 10 min, and then 40 cycles of
92°C for 15 sec and 60°C for 1 min in a GeneAmp 9700 system.
All 96-well plates contained 80 samples of unknown genotype, six known allele 1
homozygotes, six known allele 2 homozygotes, and four reactions with reagents
but no DNA. The homozygote and control samples without DNA were required for the
SDS 7700 signal processing that is outlined in the TaqMan Allelic Discrimination
Guide (PE Biosystems). Direct sequencing, single-stand conformation polymorphism
(SSCP), or denaturing high pressure liquid chromatography were used to confirm
control sample genotypes. PCR plates were read on the ABI 7700 instrument in the
end-point analysis mode of the SDS version 1.6.3 software package (ABI).
Genotypes were visually determined by comparison with the dye-component
fluorescent emission data shown in the X-Y scatter-plot of the SDS software.
Genotypes were also automatically determined by the signal processing algorithms
in the software. Results of both scoring methods were saved to two output files
for later comparison.
|
|
HAPLOTYPE-BASED
CASE-CONTROL STUDY |
|---|
We performed haplotype analysis on the 4 SNPs. Based on the genotype data of the 4 genetic variations, the frequency of each haplotype was estimated using the expectation/maximization (EM) algorithm [14,15]. For the haplotype-based case-control study determinations, SNPAlyze version 3.2 was used (Dynacom Co., Ltd., Yokohama, Japan), which is available from their website at http://www.dynacom.co.jp/products/package/snpalyze/index.html.
|
|
STATISTICAL
ANALYSIS |
|---|
Data are shown as mean ±SD. Differences between the EH and NT groups were assessed by analysis of variance (ANOVA) followed by a Fisher's protected least significant difference (PLSD) test. Hardy-Weinberg equilibrium was assessed by a chi-square analysis. When the sizes of the expected values were small (below 2.0), the genotypes were combined [16]. The overall distribution of the SNP alleles was analyzed by 2×2 contingency tables, and the distribution of the SNP genotypes between the EH patients and NT controls was tested using a 2-sided Fisher exact test and multiple logistic regression analysis. Statistical significance was established at p < 0.05. The threshold value of the frequencies of the haplotypes included in the analysis was set to 1/2n (n: numbers of subjects in each group), as suggested by Excoffier and Slatkin [17]. All haplotypes below the threshold value were excluded from the analysis. Overall distribution of haplotypes was analyzed using 2 ×m contingency tables with a value of p < 0.05 considered to indicate statistical significance. The p value significance of each haplotype was determined by the chi-square analysis and permutation method using the software SNPAlyze version 3.2 [16].
|
|
RESULTS |
|---|
|
|
|
|
Table 1 shows the clinical features for the EH patients and the NT
controls. The SBP and DBP were significantly higher in the EH group as
compared to that seen in the NT group. Age, body mass index (BMI), pulse
rate, serum concentrations of creatinine and uric acid, and plasma
concentrations of total cholesterol did not significantly differ between
the two groups.
We performed an association study using 4 SNPs. Table 2 shows the genotype
distributions of the 4 SNP variants and allelic frequencies. The overall
distribution of the genotypes did not significantly differ between the
EH and NT groups. However, there was a difference noted for the allelic
frequency of the SNP rs2290105 between women in the EH and NT groups (p
value = 0.0409), with the allelic frequency of the minor allele in the
EH group higher than that seen for the NT group.
The haplotype-based case-control study also documented significant
differences between the women of the EH and NT groups (Table 3). There
were significant differences for the results of the A-T-A-C haplotype,
with this haplotype higher in the EH (42.6%) than in the NT group
(28.8%).
| |
|---|
APA cleaves the N-terminal aspartyl acid residue of Ang II to produce Ang
III. An accumulation of Ang II leads to an increase in blood pressure. A
previous animal study that examined the APA deficient mouse documented a
relationship between hypertension and the APA gene [1] That study indicated that
the SBP and DBP of the APA-/- mouse were significantly higher than those seen in
the APA+/- mouse and the APA+/+ mouse. Therefore, the metabolism of Ang II by
APA is an important factor in the regulation of blood pressure [1-5,18,19].
This suggests that the APA gene might be an indicator of disease susceptibility
for EH. The use of SNPs is a valuable tool for association studies examining
genomic markers. Since the SNPs consisted of 3 genotypes and 2 alleles, we were
able to investigate the association between EH and the frequency of the genotype
and the allele. We examined the association between the APA gene and EH using 4
SNPs of this gene. As there have been no previous association studies concerning
the EH and APA gene, our study is the first one to attempt to determine this
relationship.
In the present study, we found no differences in genotypic frequency between the
EH and control groups. However, the allelic frequency of rs2290105 was
significantly different between the women in the EH and NT groups. There have
been previous reports on the APA knockout mouse, but these studies did not
divide the mice into sexual groups [1]. Therefore our data
suggest that SNP rs2290105 might be a genetic marker for EH in women.
Genetic association studies have previously identified genes correlated with a
gender-specific susceptibility to essential hypertension [20,21].
However, the reason for the present finding of a positive association between EH
and SNP rs2290105 in women is unclear. There are other studies that have
examined the association of the components of the renin-angiotensin system with
gender-specific hypertension [22,23].
These observations highlight the potential importance of gender-dependent
interactions between the genetic background and the expression of the
hypertensive phenotype.
In conclusion, the present study is the first to examine the correlation between
the human APA gene and EH. The present data indicate that the APA gene is a
promising candidate for use as a genetic marker for EH in women.
|
|
ACKNOWLEDGMENTS |
|---|
We would like to thank Mr. N. Sato and Ms. K. Sugama for their excellent technical assistance. This work was supported by a grant from the Ministry of Education, Science and Culture of Japan (High-Tech Research Center, Nihon University), a research grant from the alumni association of Nihon University School of Medicine, and a grant from the Tanabe Biomedical Conference, Japan.
|
|
REFERENCES
|
|---|