| Original Article |
|
|
1Department of Biochemistry, All India Institute of Medical Sciences, New Delhi, India
Corresponding author:Neeta Singh, Ph.D, Room No. 3027A, Department of Biochemistry, All India Institute of Medical Sciences, New Delhi1-110029, India. Tel: 91-11-26594945; Fax: 91-11-26588663; E-mail: singh_neeta@hotmail.com.
| |
ABSTRACT |
|
|
INTRODUCTION |
|
|
MATERIAL AND METHODS |
|
|
RESULTS |
|
|
DISCUSSION |
|
|
ACKNOWLEDGEMENTS |
|
|
REFERENCES |
|
|
ABSTRACT
|
|---|
Recently, many studies have shown that celecoxib induces apoptosis in various cancer cells by different mechanisms depending on the cell type. This study examined the effect of the selective COX-2 inhibitor celecoxib on cisplatin and TNF-α cytotoxicity and studied the role of mitochondria in the induction of apoptosis in the human cervical carcinoma SiHa cells. Apoptosis was detected by flow cytometry. The protein expression of Bcl-2, Bcl-XL, Bax, cytochrome c and AIF was analyzed by Western Blotting. The mRNA level of anti-oxidant enzymes was quantitated by RT-PCR. Priming SiHa cells with celecoxib increased the cisplatin-induced apoptosis by 20.56% and priming with celecoxib increased the TNF-α induced apoptosis by 22.07%. This was accompanied by downregulation of Bcl-XL and Bcl-2 and upregulation of Bax. Cytosolic cytochrome c increased by 43.0% with celecoxib and TNF-α treatment but was not significant with celecoxib and cisplatin treatment. Nuclear AIF increased by 21.0% with celecoxib and cisplatin treatment whereas it was not significant with celecoxib and TNF-α treatment. The mRNA level of Mn-Superoxide dismutase, CuZn-Superoxide dismutase, Glutathione peroxidase and Catalase decreased significantly on priming with celecoxib and then treating with cisplatin or TNF-α. There was no significant increase in the activity of caspase-3 with either celecoxib or TNF-α treatment or with celecoxib and cisplatin treatment. The findings suggest that priming with celecoxib induces the TNF-α and cisplatin-mediated apoptosis in SiHa cells perhaps through ROS-mediated mitochondrial pathway.
KEY WORDS:
cervical cancer; apoptosis celecoxib; cisplatin; TNF-α; mitochondria
|
|
INTRODUCTION |
|---|
Cervical cancer is one of the most common cancers amongst women. Despite its frequency and recurrence, the death rate has been declining over the past 40 years, due to early detection and treatment (1). Currently, the standard therapy for cervical carcinoma, following adequate radical surgery is simultaneous radiochemotherapy with a platinous chemotherapeutic agent. However, one disadvantage is that a number of patients have to break off therapy because of treatment-related toxicities (2). Cisplatin is among the most widely used and most effective chemotherapeutic agent for many types of human cancers. Because killing cancer cells by chemotherapy is principally executed by apoptosis, a detailed understanding of the apoptotic program and the factors that might influence cell death are essential for designing novel and more effective drug targets with relatively lesser side effects.
Many natural and synthetic compounds have been shown to modulate cisplatin and TNF-α mediated cytotoxicity in human cervical carcinoma cells. Vitamin C has been shown to enhance cisplatin-mediated apoptosis in human cervical carcinoma cells by downregulating transcription factor AP-1 and stabilizing P53 (3). Tumor necrosis factor-alpha activates both cell death and cell survival pathways, which render most cancer cells resistant to its cytotoxicity. Luteolin, a plant flavonoid, greatly sensitized TNF-α induced apoptotic cell death in a number of human cancer cell lines including cervical cancer cells by inhibiting TNF-α induced activation of nuclear transcription factor-kappa B, the main survival factor in TNF-α signaling (4).
The enzyme cyclooxygenase-2 (COX-2) has been associated with carcinogenesis and overexpression of COX-2 protein has been reported in a number of cancers including hepato-cellularcarcinoma (5), gastric cancer (6), esophageal cancer (7) colon cancer (8) and cervical carcinoma (9). COX-2 is also involved in apoptosis resistance, angiogenesis, and tumor cell invasiveness, which appear to contribute to its effects in tumorigenesis (10). Its relationship to apoptosis in cervical carcinoma patients treated with neoadjuvant chemotherapy has important clinical implications. An increased expression of COX-2 is associated with poor response to chemotherapy (11).
Celecoxib is a selective cyclooxygenase-2 (COX-2) inhibitor and non-steroidal anti-inflammatory drug (NSAID), which has been shown to be capable of inhibiting the growth of various cancer cell lines. This growth inhibitory effect was found to be independent of the COX-2 protein expression level of the cells lines (12). In the present study we investigated the enhancement of cytotoxic effects of cisplatin and TNF-α by selective COX-2 inhibitor celecoxib on human cervical cancer cells. We also investigated the role of mitochondrial pathway of apoptosis in the celecoxib-enhanced cytotoxic effects of cisplatin and TNF-α.
|
|
MATERIAL AND METHODS
|
|---|
Cell Culture and Treatments
Human Cervical Carcinoma cells SiHa were grown in DMEM medium supplemented with 10% fetal calf serum and containing antibiotics (100 U/ml penicillin, 0.1 mg/ml streptomycin) in a humidified atmosphere of 5% CO2 in air at 370C. Logarithmically growing cells were used for all experiments. Cells were treated with standardized doses of celecoxib (100μM) for 24 hours followed by cisplatin (5μg/ml) for another 24 hours. Cells were also treated with celecoxib (100μM) for 24 hours followed by TNF-α (5ng/ml) for another 24 hours.
Isolation of Nuclei, Mitochondria and Cytosol
Cells were sonicated in buffer containing 10 mM Tris-HCl pH 7.5, 10 mM NaCl and 1.5mM MgCl2, 175mM Sucrose and 12.5mM EDTA and the cell extract centrifuged at 1000g for 10 minutes to pellet the nuclei. The supernatant thus obtained was centrifuged at 18,000g for 30 minutes to pellet the mitochondria. The mitochondria were purified as described (13). The resulting supernatant was termed as the cytosolic fraction. The pellets were lysed and protein was estimated in all the three fractions by Bradford’s method (14). The purity of the fractions was confirmed by assaying the marker enzymes succinate dehydrogenase for mitochondria, lactate dehydrogenase for the cytosol and NMN adenylyl transferase for the nuclear fraction.
The whole cells, nuclei or the mitochondrial fractions were lysed in ice-cold lysis buffer (50mM Tris, 150mM NaCl, 2.5mM EDTA, 0.1% SDS, 0.5% sodium deoxycholate, 1% NP-40 and 0.02% sodium azide) containing protease inhibitors as described (15). After incubation for 30 minutes on ice, samples were centrifuged at 10,000 rpm for 30 minutes. Protein content in the lysed extracts was determined using the Bradford’s method. Equal amounts of protein (80 µg/lane) was loaded on 10-12% SDS-polyacrylamide gels and transferred to nitrocellulose membranes. The primary antibodies against Bcl-XL, Bcl-2, Bax, cytochrome c and AIF were obtained from Santa Cruz Biotechnology USA. The secondary antibody used was the appropriate alkaline phosphatase conjugated antimouse, antirabbit or antigoat IgG (Promega, Madison, WI). Proteins were detected using BCIP-NBT substrate from Promega. Densitometric scanning was performed using Alpha Imager 2200. Positive and negative controls were run with each antibody.
Flow Cytometry
Apoptosis was measured by flow cytometry as described earlier (16). In brief the cells were washed twice with PBS and fixed in 70% ethanol overnight. Cells were then washed twice with PBS to remove ethanol and then incubated in propidium iodide (20ug/ml) for 1 hour. Flow cytometry was performed using an EPICS XL-MCL flow cytometer (Coulter Electronics,Miami,FL) software. Win MDI 2.8 Software was used to generate histograms, which were then used to determine the cell cycle phase distribution after debris exclusion. The sub G1-G0 cell fraction was considered as representative of apoptotic cells.
Reverse Transcriptase mediated-PCR
The mRNA levels of various enzymes related to oxidative stress were analyzed by RT-PCR. RNA was isolated using TRI-Reagent from Sigma and precipitated by isopropanol as described earlier (17). The precipitated RNA was dissolved in DEPC-water. The purity of the isolated RNA was checked by running it on formaldehyde gel and quantitated by taking absorbance at 260nm and 280nm. The RT –Reaction mixture was incubated at 420C for 10 min to disrupt the secondary structure in the template of RNA.This was followed by the addition of reverse transcriptase enzyme from Stratagene and incubation at 370C for 60 min for the synthesis of cDNA, which was then used as a template for PCR using primers specific for Mn-Superoxide Dismutase, Cu-Zn Superoxide Dismutase, Glutathione Peroxidase and Catalase mRNA.
Forward primer: 5’ GGCCTGATTATCTAAAAGCTATTTGG 3’
Reverse primer: 5’ CGATCGTGGTTTACTTTTTGCA 3’
Copper-Zinc superoxide dismutase
Forward primer: 5’ TGGTGGTCCATGAAAAAGCA 3’
Reverse primer: 5’ CCAGCGTTTCCTGTCTTTGTACT 3’
Glutathione peroxidase
Forward primer: 5’ TGTGCCCCTACGGAGGTAC 3’
Reverse primer: 5’ AGCTGGGCCCTTGAGACAG 3’
Catalase
Forward primer: 5’ AGAGGAAACGTCTGTGTGAGAACA 3’
Reverse primer: 5’ TGACCGCTTTCTTCTGGATGA 3’
Bcl-2
Forward primer: 5’ GCCGGTTCAGGTACTCAGTCA 3’
Reverse primer: 5’ CATGTGTGTGGAGAGCGTCAA3’
Caspase-3 activity was measured by the direct assay of enzyme activity in cell lysates using synthetic fluorogenic substrate Ac-DEVD-AMC. Cells were washed with PBS, pH 7.5 and lysed in 10 mM Tris HCl, pH 7.5, 130 mM NaCl, 1% Triton X-100, 10 mM Na4P2O7 and 10 mM, Na2HPO4 on ice (10 × 106 cells / ml of lysis buffer). 100µl of cell lysate was added to the reaction buffer (20 mM HEPES, pH 7.2; 10% Glycerol; 2 mM DTT; 250 µM Ac-DEVD-AMC) and incubated at 37°C for one hour. Amounts of fluorogenic moiety released were measured using a spectrofluorimeter with excitation at 380 nm and emission at 420-460 nm (18).
Each experiment was repeated three times. All data are expressed as mean ± SD. Students t test was used to determine the significance between the control and various experimental groups. A difference was considered statistically significant at p < 0.05.
|
|
RESULTS
|
|---|
Celecoxib enhanced cisplatin and TNF- α induced cell death in SiHa cells
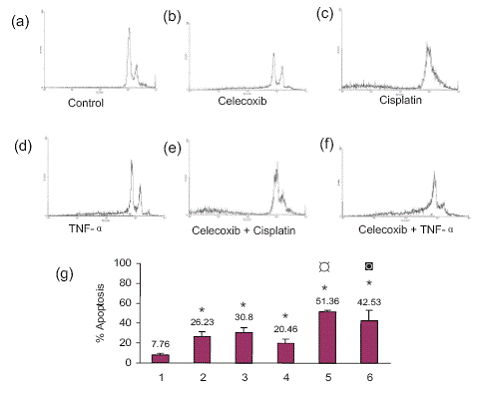
Apoptosis was observed by morphological changes such as cell shrinkage, rounding of the cells and formation of apoptotic bodies. This was more prominent for the combination treatments (Figure 1). The untreated control cells showed 7.76% apoptosis. Celecoxib by itself caused 26.23% apoptosis, cisplatin by itself induced 30.80% apoptosis and TNF-α by itself induced 20.46% apoptosis. Pretreatment with celecoxib increased cisplatin-induced apoptosis to 51.36% and that of TNF-α to 42.53% (Figure 2). The morphological changes were in agreement with the flow cytometry data.
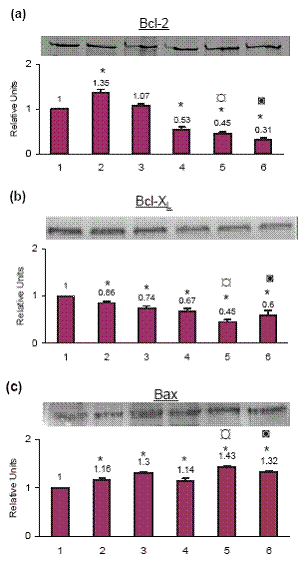
Bcl-2 was downregulated by celecoxib pretreatment
Celecoxib treatment by itself increased Bcl-2 protein level by 35.0% as compared to the control cells. Cisplatin treatment increased Bcl-2 by 7.0% (not significant) and TNF-α by 47.0% as compared to the control cells. On priming with celecoxib followed by treatment with cisplatin the Bcl-2 decreased by 55.0% and by 69.0% on TNF-α treatment as compared to the control cells (Fig. 3a).
Bcl-XL was downregulated by celecoxib pretreatment
By itself celecoxib treatment decreased Bcl-XL by 14.0%, cisplatin by 26.0% and TNF-α by 33.0% as compared to the control cells. When cells were primed with celecoxib and then treated with cisplatin the Bcl-XL level decreased by 55.0% whereas when treated with TNF-α the Bcl-XL decreased by 40.0% (Fig. 3b).
Bax was upregulated by celecoxib pretreatment
Celecoxib treatment by itself increased Bax by 16.0%, cisplatin treatment increased Bax by 30.0% and TNF- α treatment by 14.0% as compared to the control cells. When cells were primed with celecoxib and then treated with cisplatin the Bax increased by 43.0% and with TNF-α it increased by 32.0% as compared to the control cells (Fig. 3c).
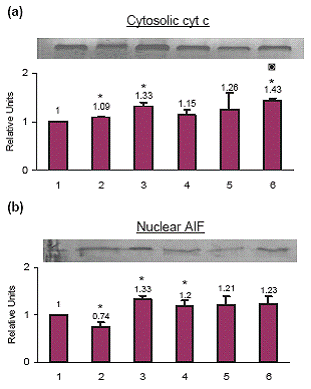
No Translocation of cytochrome c to the cytosol by celecoxib pretreatment
Treatment with celecoxib increased cytosolic cytochrome c by 9.0%, cisplatin by 33.0% and TNF-α by 15.0% as compared to the control cells. When cells were primed with celecoxib and then treated with cisplatin the cytosolic cytochrome c increased by 26.0% whereas when cells were primed with celecoxib and then treated with TNF-α the cytosolic cytochrome c increased by 43.0% as compared to the control cells (Fig. 4a).
AIF is not involved in the celecoxib- or TNF-mediated apoptosis
Celecoxib treatment decreased nuclear AIF by 26.0%, cisplatin treatment increased nuclear AIF by 33.0% and TNF-α treatment increased nuclear AIF by 20.0% as compared to the control cells, which was not significant. When cells were primed with celecoxib and then treated with cisplatin the nuclear AIF increased by 21.0% whereas when cells were primed with celecoxib and treated with TNF-α the nuclear AIF increased by 23.0% as compared to the control cells which was not significant (Fig. 4b).
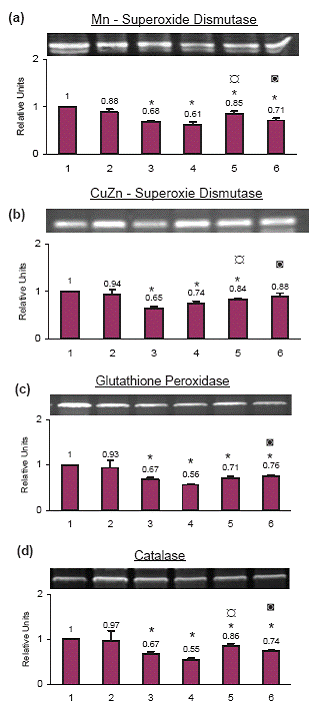
Anti-oxidant enzymes at the mRNA level
Mn-Superoxide dismutase. The mRNA level of Mn-Superoxide dismutase decreased significantly with cisplatin (34.0%), TNF-α (39.0%), celecoxib+cisplatin (15.0%) and celecoxib + TNF- α (29.0%) treatment. The decrease with celecoxib alone (12.0%) was not significant as compared to the control cells (Fig. 5a).
CuZn-Superoxide dismutase. The mRNA level of CuZn-Superoxide dismutase decreased by 35.0% with cisplatin treatment alone, 26.0% with TNF-α treatment alone and 16.0% with celecoxib+cisplatin treatment. The change with celecoxib alone or with celecoxib+ TNF- α treatment was not significant as compared to the control cells (Fig. 5b).
Glutathione peroxidase. Glutathione peroxidase mRNA decreased significantly with cisplatin (33.0%), TNF-α (44.0%), celecoxib+cisplatin (29.0%) and celecoxib+ TNF- α (24.0%) as compared to the control cells. The increase with celecoxib alone (7.0%) was not significant as compared to the control cells (Fig. 5c).
Catalase. Catalase mRNA decreased significantly with cisplatin (33.0%), TNF-α (45.0%), celecoxib+cisplatin (14.0%) and celecoxib+ TNF-α (26.0%) as compared to the control cells. The decrease with celecoxib alone (3.0%) was not significant as compared to the control cells (Fig. 5d).
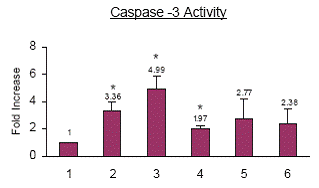
Celecoxib priming does not result in caspase-3 activation. Celecoxib treatment increased caspase-3 activity by 3.36 fold, cisplatin treatment increased caspase-3 activity by 4.99 fold and TNF-α treatment increased caspase-3 activity by 1.97 fold as compared to the control cells. When cells were primed with celecoxib and then treated with cisplatin, caspase-3 activity increased by 2.77 fold as compared to the control cells which was not significant. Whereas when cells were primed with celecoxib and treated with TNF-α caspase-3 activity increased by 2.38 fold, as compared to the control cells which were not significant (Fig. 6).
|
|
DISCUSSION
|
|---|
Cervical carcinoma is a human papillomavirus (HPV)-associated cancer for which treatment options still mainly rely on surgical procedures, with or without adjuvant radiotherapy and chemotherapy (19). Recent advances in cancer chemotherapy have drawn the attention of investigators to the usefulness of chemotherapy for cancer of the uterine cervix. Though satisfactory therapeutic results have been achieved with cisplatin-based cyclic balloon-occluded arterial infusion chemotherapy (BOAI), nevertheless, there have been some patients in whom it was ineffective and these patients showed significantly higher expression of COX-2 after BOAI, accompanied by cancer cell apoptosis inhibition. It has been suggested that overexpression of COX-2 inhibits cancer cell apoptosis and adversely influences the prognosis (20). Thus assessment of the COX-2 status could be useful to identify cervical cancer patients who may benefit from neoadjuvant chemotherapy (21). In cervical cancer, celecoxib treatment decreases tumor COX-2 expression and markers of proliferation and neoangiogenesis, thus suggesting that selective COX-2 inhibitors may be a promising strategy not only for chemopreventive approaches but also for therapeutic approaches in cervical cancer (22).
Tumor necrosis factor- alpha (TNF-α) induces apoptosis of a variety of tumor cell types. Celecoxib induces apoptosis independent of COX-2 activity. Caspase-8 and -9 are implicated in the apoptotic effect of celecoxib in cervical cancer cells. We investigated the effect of the selective COX-2 inhibitor celecoxib on cisplatin and TNF-α cytotoxicity and studied the role of the mitochondrial pathway in the induction of apoptosis in SiHa cells. In the present study a 100 μM dose of celecoxib induced significant apoptosis in SiHa cells and it enhanced both the cisplatin and TNF-α mediated apoptosis. This was accompanied by a decrease in the expression of Bcl-2 and Bcl-XL and an increase in the Bax protein levels. Bcl-2 and Bcl-XL exert their anti-apoptotic effect, at least in part by binding to Bax and related pro-apoptotic proteins. A higher expression of the Bax protein and gene has been associated with accelerated cancer cell apoptosis. On the other hand, the patients in whom cisplatin-based therapy was ineffective showed higher expression of the Bcl-XL protein and gene (23).
Because Bcl-2 family proteins can regulate release of cytochrome c and AIF from the mitochondria, the proteins Bcl-2 and Bcl-XL play a significant role in the process of induction of apoptosis. Cytochrome c released from the mitochondria forms a complex with procaspase-9 and apoptotic protease-activating factor-1 (Apaf-1), resulting in activation of procaspase-9. In contrast, AIF is believed to be responsible for chromatin condensation and is usually associated with caspase-independent cell death. Similar to cytochrome c, AIF is released from the mitochondria in response to death stimuli. On induction of apoptosis, AIF is translocated to the nucleus and causes large-scale DNA fragmentation and chromatin condensation in a caspase-independent manner (24). There was no significant change in the cytosolic cytochrome c and nuclear AIF levels with the combination treatment of celecoxib and cisplatin. Further the caspase-3 activity was also not significantly increased.
There was a significant increase in the cytosolic cytochrome c (43.0%) with the combination treatment of celecoxib and TNF. This observed increased cytochrome c release accompanied but non significant increase in caspase-3 activity implies that cytochrome c does not mediate further downstream events in the apoptotic pathway. Probably there is an inhibition at the level of caspase activation by Inhibitor of Apoptosis Proteins (IAPs). Nuclear AIF was not significantly altered with the combination treatment of celecoxib and TNF-α implicating the non-involvement of AIF in the apoptotic process.
Mitochondria are considered to be a major source of intracellular oxygen radicals. Accumulation of ROS results in cellular oxidative stress, and if not corrected, can lead to the damage of important biomolecules such as membrane lipids, proteins and DNA and its prolonged accumulation may cause irreversible cellular injury ultimately resulting in cell death. The mRNA levels of various enzymes related to oxidative stress showed significant decrease suggesting the role of oxygen radicals in the induction of apoptosis. Given the substantial role of mitochondria in ATP metabolism, in generation of free radicals, and in the regulation of apoptosis, mitochondrial dysfunction seems likely to affect cellular energy capacities, increase oxidative stress, cause ROS-mediated damage to DNA and alter the cellular response to apoptosis induction by anticancer agents.
Arsenic trioxide is known to induce apoptosis in human cervical cancer cells through a reactive oxygen species-dependent pathway involving loss of the mitochondria membrane permeability (∆ m) and caspase-3 activation (25). Precise mechanism of the ROS-mediated caspase-independent apoptotic cell death triggered by As2O3 treatment still remains unclear.
These results are consistent with previous reports that Bcl-2 can block loss of mitochondria membrane permeability and AIF release (26). In some systems, Bcl-2 (or Bcl-XL) appears to influence ROS generation (27) whereas in others it does not affect ROS production but does prevent oxidative damage to cellular constituents (28). It has also been proposed that Bcl-2 modulates level of ROS through induction of intracellular antioxidants. This activity of Bcl-2 may control entry into apoptosis (29).
Caspase activation is thought to be an important mechanism for the apoptotic cell death program; however, our findings suggest that cell death induced by the combination treatment of celecoxib and TNF-α in human cervical cancer cells is caspase-independent as there was no significant increase in of caspase-3 activity though the cytosolic cytochrome c was elevated. There appears to be a block at the level of caspase inhibition possibly by IAPs. Neither there was any significant change in the cytosolic cytochrome c and the caspase-3 activity with celecoxib and cisplatin treatment. Possibly a reactive oxygen-mediated caspase-independent pathway is involved. Such a mechanism of cell death has been recently reported in human cervical cancer cells (30).
|
|
|
|
|
|
|
|
ACKNOWLEDGEMENTS
|
|---|
The authors acknowledge Indian Council of Medical Research and Department of Biotechnology, Govt. of India for financial support.
|
|
REFERENCES
|
|---|
1. Shehata M, Shehata M, Shehata F, Pater A. Dual apoptotic effect of Xrel3 c-Rel/NF-kappaB homolog in human cervical cancer cells. Cell Biol Int. 2004; 28: 895-904.
2. Kummel S, Thomas A, Jeschke S, Hauschild M, et al. Postoperative therapy modalities for cervical carcinoma. Anticancer Res. 2006; 26: 1707-1713.
3. Reddy VG, Khanna N, Singh N. Vitamin C augments chemotherapeutic response of cervical carcinoma HeLa cells by stabilizing P53. Biochem Biophys Res Commun. 2001; 282: 409-415.
4. Shi RX, Ong CN, Shen HM. Luteolin sensitizes tumor necrosis factoralpha- induced apoptosis in human tumor cells. Oncogene. 2004; 23: 7712-7721.
5. Chi-Man Tang T, Tung-Ping Poon R, Fan ST. The significance of cyclooxygenase- 2 expression in human hepatocellular carcinoma. Biomed Pharmacother. 2005; 59 Suppl 2: S311-316.
6. Zhang J, Ji J, Yuan F, Zhu L, et al. Cyclooxygenase-2 expression is associated with VEGF-C and lymph node metastases in gastric cancer patients. Biomed Pharmacother. 2005; 59 Suppl 2: S285-288.
7. Shimizu D, Vallbohmer D, Kuramochi H, Uchida K, et al. Increasing cyclooxygenase-2 (cox-2) gene expression in the progression of Barrett's esophagus to adenocarcinoma correlates with that of Bcl-2. Int J Cancer. 2006; 119: 765-770.
8. Strillacci A, Griffoni C, Spisni E, Manara MC, et al. RNA interference as a key to knockdown overexpressed cyclooxygenase-2 gene in tumour cells. Br J Cancer. 2006; 94: 1300-1310.
9. Dai Y, Zhang X, Peng Y, Wang Z. The expression of cyclooxygenase- 2, VEGF and PGs in CIN and cervical carcinoma. Gynecol Oncol. 2005; 97: 96-103.
10. Sinicrope FA. Targeting cyclooxygenase-2 for prevention and therapy of colorectal cancer. Mol Carcinog. 2006; 45. 447-454.
11. Ishiko O, Sumi T, Yoshida H, Matsumoto Y, et al. Association between overexpression of cyclooxygenase-2 and suppression of apoptosis in advanced cancer of the uterine cervix after cyclic balloon-occluded arterial infusion. Oncol Rep. 2001; 8: 1259-1263.
12. Chan CM, Ma BB, Wong SC, Chan AT. Celecoxib induces dose dependent growth inhibition in nasopharyngeal carcinoma cell lines independent of cyclooxygenase-2 expression. Biomed Pharmacother. 2005; 59 Suppl 2: S268-271.
13. Gui-Qiang Wang, Brian R. Gastman, Eva Wieckowski, Goldstein, Andrea Gambotto, Tae-Hyoung Kim, Bingliang Fang, Asaf Rabinovitz, Xiao-Ming Yin, and Hannah Rabinowich, et al.. A Role for Mitochondrial Bak in Apoptotic Response to Anticancer drugs. J Biol Chem. 2001; 276: 34307-34317.
14. Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976; 72: 248-254.
15. Sharma H, Sen S, Mathur M, Bahadur S, et al. Combined evaluation of expression of telomerase, survivin, and anti-apoptotic Bcl-2 family members in relation to loss of differentiation and apoptosis in human head and neck cancers. Head Neck. 2004; 8: 733-740.
16. Khanna N, Reddy VG, Tuteja N, Singh N. Differential gene expression in apoptosis: identification of ribosomal protein S29 as an apoptotic inducer. Biochem Biophys Res Commun. 2000; 277: 476-486.
17. Sharma H, Sen S, Muzio LL, Mariggio A, et al. Antisense-mediated downregulation of anti-apoptotic proteins induces apoptosis and sensitizes Head and Neck Squamous Cell Carcinoma Cells to Chemotherapy. Cancer Biol Ther. 2005; 4: 720-727.
18. Sen S, Sharma H, Singh N. Curcumin enhances Vinorelbine mediated apoptosis in NSCLC cells by the mitochondrial pathway. Biochem Biophys Res Commun. 2005; 331: 1245-1252.
19. Simonart T, Boelaert JR, Andrei G, Clercq ED, et al. Iron withdrawal strategies fail to prevent the growth of SiHa-induced tumors in mice. Gynecol Oncol. 2003; 90: 91-95.
20. Ishiko O, Sumi T, Yoshida H, Matsumoto Y, et al. Association between overexpression of cyclooxygenase-2 and suppression of apoptosis in advanced cancer of the uterine cervix after cyclic balloon-occluded arterial infusion. Oncol Rep. 2001; 8: 1259-1263.
21. Nagai N, Tian X, Mukai K, Hirata E, et al. Overexpression of cyclooxygenase- 2 protein and its relationship to apoptosis in cervical carcinoma treated with neoadjuvant chemotherapy. Int J Mol Med. 2003; 12: 709-714.
22. Ferrandina G, Ranelletti FO, Legge F, Lauriola L, et al. Celecoxib modulates the expression of cyclooxygenase-2, ki67, apoptosis-related marker, and microvessel density in human cervical cancer: a pilot study. Clin Cancer Res. 2003; 9: 4324-4331.
23. Li P, Nijhawan D, Budihardjo I, et al. Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell. 1997; 91: 479-489.
24. Fisher, T.C., Milner,A.E., Gregory,C.D., Jackman,A.L., et al. Bcl-2 modulation of apoptosis induced by anticancer drugs: resistance to thymidylate stress is independent of classical resistance pathways. Cancer Res. 1993; 53: 3321–3326.
25. Woo SH, Park IC, Park MJ, Lee HC, et al. Arsenic trioxide induces apoptosis through a reactive oxygen species-dependent pathway and loss of mitochondrial membrane potential in HeLa cells. Int J Oncol. 2002; 21: 57-63.
26. Kluck RM, Bossy-Wetzel E, Green DR, Newmeyer DD. The release of cytochrome c from mitochondria: a primary site for Bcl-2 regulation of apoptosis. Science. 1997; 275: 1132-1136.
27. Gottlieb E, Vander Heiden MG, Thompson CB. Bcl-x (L) prevents the initial decrease in mitochondrial membrane potential and subsequent reactive oxygen species production during tumor necrosis factoralpha- induced apoptosis. Mol Cell Biol. 2000; 20: 5680-5689.
28. Tyurina YY, Tyurin VA, Carta G, Quinn PJ, et al. Direct evidence for antioxidant effect of Bcl-2 in PC12 rat pheochromocytoma cells. Arch Biochem Biophys. 1997; 344: 413-423.
29. Steinman HM. The Bcl-2 oncoprotein functions as a pro-oxidant. J Biol Chem. 1995; 270: 3487-3490.
30. Kang YH, Yi MJ, Kim MJ, Park MT,et al. Caspase-independent cell death by arsenic trioxide in human cervical cancer cells: reactive oxygen species-mediated poly (ADP-ribose) polymerase-1 activation signals apoptosis-inducing factor release from mitochondria. Cancer Res. 2004; 64: 8960-8967.