| ORIGINAL ARTICLE |
|
|
1 Department of Pharmacy, College of Jiangsu Jiankang Profession, Nanjing, P. R. China;
2 Department of Pharmacy, NanJing University of Chinese Medicine, Nanjing, P. R. China
Corresponding Author: Haijun Yang, Department of Pharmacy, College of Jiangsu Jiankang Profession, Nanjing 210029, P. R. China. Tel: +86 25 8581 1521; Fax: 86 25 8581 1563; E-mail: yanghaijunzn@163.com.
| |
ABSTRACT |
| INTRODUCTION | |
|
|
EXPERIMENTAL |
|
|
RESULTS AND DISCUSSION |
|
|
CONCLUSIONS |
|
|
ACKNOWLEDGEMENT |
|
|
REFERENCES |
|
|
ABSTRACT
|
|---|
Annonaceous acetogenins (ACGs) isolated from Annonaceaeplants exhibited a broad range of biological bioactivities such as cytotoxic, antitumoral, antiparasitic, pesticidal and immunosuppresive activities. However, their structures were liable to change at more than 60°C and their extraction yields were low using traditional organic solvent extraction. In the present study, all samples from Annona genus plant seeds were extracted by supercritical carbon dioxide under optimized conditions and a high-performance liquid chromatography (HPLC) method was established for simultaneously determining ten ACGs. All of the ten compounds were simultaneously separated on reversed-phase C18 column (250 mm × 4.6 mm, 5 μm) with the column temperature at 30°C. The mobile phase was composed of (A) methanol and (B) distilled water, the flow rate was 1.0 ml/min and the detection wavelength was set at 220 nm. All calibration curves showed good linear regression (γ>0.9995) within the test range. The established method showed good precision and accuracy with overall intra-day and inter-day variations of 0.99-2.56% and 1.93-3.65%, respectively, and overall recoveries of 95.16-105.01% for the ten compounds analyzed. The established method can be applied to evaluate the intrinsic quality of Annonaceae plant seeds. The determination results recover the content-variation regularities of various ACGs in different species, which are helpful to choose the good-quality Annonaceae plant seeds for anticancer lead compound discovery.
KEY WORDS: Annonaceae plant seeds; supercritical fluid CO2 extraction; quantification; HPLC–DAD; annonaceous acetogenins|
|
INTRODUCTION |
|---|
Annonaceous acetogenins (ACGs) constitute a series of natural products isolated exclusively from Annonaceae plants (1–4), which are comprised of some 130 genera and include over 2300 species and are widely distributed in tropical and sub-tropical regions. More than 500 ACGs have been isolated and identified from this plant family, mostly from the seeds and stem bark. Chemically, the annonaceous acetogenins are white, waxy, derivatives of long-chain (C35 or C37) fatty acids. They are usually character- ized by a long aliphatic chain bearing a terminal methyl-substituted α, β -unsaturated γ - lactone ring with one, two, three tetrahydrofuran (THF) or tetrahydropyran (THP) rings. ACGs exhibited a broad range of biological activities such as cytotoxic, antitumoral, antiparasitic, pesticidal and immunosuppresive activiti- es (5-10). Especially, their ability to inhibit multiple drug resistant (MDR) tumor cell lines (11, 12) has attracted much attention of chemists and biologists. The ACGs are the most powerful known inhibitors of complex I (NADH: biquinone oxidoreductase) in mammalian and insect mitochondrial electron transport system (13–15). In addition, they are potential inhibitors of NADH oxidase of the plasma membranes of cancer cells, the inhibition results in a depletion of ATP levels which causes arrest in the cell cycle at the G1 phase, and subsequently apoptosis is induced (16-20). So, ACGs are regarded as a likely source for the development of potential drugs.
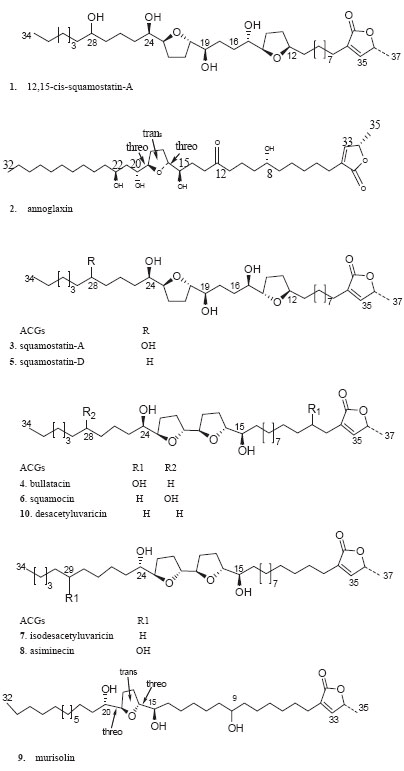
However, ACGs are low polarity compounds and their structures were liable to change at more than 60ºC and their extraction yields were low using classical organic solvent extraction method. As an alternative of traditional extraction method, supercritical fluid CO2 extraction (SFE), an extraction technique under low temperature, has recently used in the extraction of bioactive constituents from herbal medicines for its small amount of solvent consumption, automated sample handling and high extractive efficiency. So in the present study all samples were extracted by SFE and the optimal technology was concluded by our previous research (21). To our knowledge, only a few analytical methods are available for determination ACGs, previously reported analytical methods were just developed to determine the total ACGs by spectrophoto- metric analysis (22), which was instable and to quantify just three types of ACGs by HPLC method (20). So in order to assess the intrinsic quality of different Annonaceae plantseeds andmeet the regulatory requirements for investigating Annonaceae plant seeds, a HPLC method was developed for simultaneous determination ten major ACGs in Annona genus plant seeds distributed in south of China. Figure 1 illustrates the representatives of the ten ACGs which represent the main structural types of bio- active ACGs and their contents are considerable in Annonaceae plantseeds.
|
|
|
EXPERIMENTAL |
|---|
Chemicals and reagents
The ten reference compounds (12,15-cis-squamostatin-A, annoglaxin, squamostatin-A, bullatacin, squamocin, squamostatin–D, isodesacetyluvaricin,asiminecin, murisolin and desacetyluvaricin) were isolated from Annon squamosa seeds by our laboratory and their structures were established based on spectroscopic analysis, the purity of each reference compound was determined to be above 99% by HPLC analysis and confirmed by LC–MS, NMR spectroscopy. HPLC-grade methanol was purchased from Hanbang Science and Technology Company (Nanjing, China), the deionized water was purified using a Milli-Q Plus 185 system from Millipore (Milford, MA, USA).
Plant material
Five Annona genus plant (Annona squamosa, A. muricata, A. glabra, A. reticulata and A. bullata) seeds were collected from Jiangsu, Hainan, Guangdong and Yunnan provinces, P. R. China and were identified by Professor Jianwei Chen (NanJing university of Chinese Medicine, Nanjing, China). After collection, the seeds were allowed to dry at ambient temperature for about one week and were then crushed and immediately extracted.
Apparatus and Chromatographic conditions
A supercritical fluid extractor SFE-2 (Applied Separation, USA) which is capable of pressure up to 680 bar and temperature up to 240°C, static and dynamic extraction with flow from 0 to 10 L/min (gaseous carbon dioxide) and extraction vessels from 5 ml to 1l were used. An Agilent 1200 liquid chromatograph system (Agilent technologies, CA, USA) consisting of binary pump, an auto-sampler and diode-array detector was used. The column configuration consisted of an Agilent Zorbax Extend reversed-phase C18 column (250 mm × 4.6 mm, 5 μm). Detection wavelength was set at 220 nm. The mobile phase consisted of A (methanol) and B (deionized water), using a linear gradient: 0-40 min (85% A), 40-60 min (85% A-95% A). The flow rate was 1.0 ml/min. The column temperature was maintained at 30°C.
Preparation of standard solutions
A mixed standard stock solution containing 12,15-cis-squamostatin-A (a), annoglaxin (b), squamostatin-A (c), bullatacin (d), squamostatin-D (e), squamocin (f), isodesacetyluvaricin (g), asiminecin (h), murisolin (i) and desacetyluvaricin (j) was prepared in methanol. Working standard solutions were prepared by diluting the mixed standard solution with methanol to give six different concentrations within the ranges: a, 3.8-45.6 μg/ml; b, 2.1-26.7μg/ml; c, 3.2-36.4 μg/ml; d, 4.5-51.7 μg/ml; e, 2.7-29.2 μg/ml; f, 0.58-17.3 μg/ml; g, 2.5-18.9 μg/ml; h, 4.3-48.9 μg/ml; i, 3.3-41.8μg/ml and j, 3.5-44.6 μg/ml for calibration curves. The standard solutions were filtered through a 0.45 μm membrane prior to injection. The standard stock and working solutions were stored at 4°C.
Preparation of sample solutions
The dried powder of Annona plant seeds (100 g, 20 mesh) was accurately weighed and extracted by SFE under optimized conditions (extraction pressure: 30 Mpa; extraction temperature: 35ºC; extraction time: one hour; 20 ml 95% ethanol modifier) (21). After evaporating ethanol to dryness by a rotary evaporator, residue was dissolved in methanol in a 25 ml flask, and then filtrated through a 0.45 micro-m millipore filter before HPLC injection. Three aliquots of the solution (20 μl) were injected to RP-HPLC-DAD system.
|
|
RESULTS AND DISCUSSION |
|---|
Optimization of separation condition
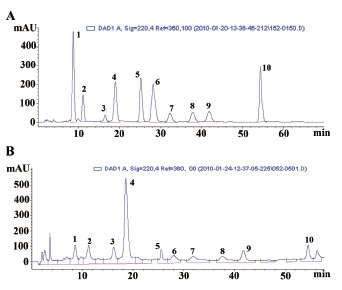
Different mobile phase compositions were examined: methanol-water and acetonitrile-water. As a result, the 85% methanol and 15% water system could give best separation of the ten reference compounds in 60 min. Furthermore, other chromatographic variables were also optimized, including analytical columns (Hanbon Hedera ODS-2, Hanbon Lichrospher C18 and Agilent Zorbax Extend C18), the colum temperatures (20ºC, 25ºC and 30ºC) and the flow rates (0.8 ml/min and 1.0 ml/min). Eventually, the optimal separation was achieved on an Agilent Zorbax Extend C18 colum (250 mm × 4.6 mm, 5 μm) at a column temperature of 30ºC with a flow rate of 1.0 ml/min. According to the absorption maxima of ten reference compounds on the UV spectra with three-dimensional chromatograms of HPLC-DAD detection, the wavelength was set at 220 nm. Representative chromatograms for the standard analytes and for a sample were shown in Fig. 2. Fig. 2A displayed that the ten standard analytes were well separated and the resolution between any two compounds was greater than 1.5. Other compounds in the sample did not interfere with analysis of the ten standard analytes, as shown in Fig. 2B. The chromatographic peaks were identified by comparing their retention time with that of each reference compound, which was eluted in parallel with the optimised mobile phases. In addition, spiking samples with the reference compounds showed no additional peaks, which further confirmed the identities of the analytes’ peaks.
|
Calibration curves, limits of detection and quantification
The calibration curves were performed with 6 different concentrations in triplicate. All calibration curves were obtained from peak areas (Y) of the standard solutions versus the concentrations for reference compounds.Linear regression analysis for each of the ten reference compounds was performed by the external standard method. The limit of detection (LOD) was determined at a signal-to-noise ratio of 3 and the limit of quantificaion (LOQ) was determined as the lowest concentration in the linear range of each analyte. The calculated results are given in Table 1. All ten reference compounds showed good linearity (r>0.9995) in a relatively wide concentration range. The limits of detection (LOD) and quantification (LOQ) of the ten analytes were 0.11-0.42 μg/ml and 0.58-4.5 μg/ml, respectively. The LOD and LOQ were reported in Table 1.
Precision and stability
Intra-day and inter-day variations were chosen to determine the precision of the developed method by analyzing certain concentrations of standard solution. For intra-day variation, the standard solution was analyzed for six times within one day, the inter-day variation was determined in three consecutive days. The overall relative standard deviations of the intra-day and inter-day were less than 3.66%. The results were shown in Table 2.
Stability study was performed with sample solution at room temperature and analyzed at 0 h, 2 h, 4 h, 8 h, 12 h, 24 h and 48 h within two days, respectively. Variations were expressed by relative standard deviations (R.S.D.). The R.S.D. of stability was not more than 4.21% for all analytes.
Recovery
An appropriate amount of sample was weighed and spiked with known amount of each standard compound. They were then treated and analyzed as described above. Each sample was analyzed in triplicate. The average recoveries were estimated by the formula: recovery (%) = (amount found – original amount)/amount spiked × 100%. The total amount of each analyte was calculated from the corresponding calibration curve. The overall recoveries lay between 95.2% and 105.1% for all reference compounds, with R.S.D. less than 4.5% indicating that the established method was accurate enough for the determination of the ten annonaceous acetogenins in Annona plant seeds. The results of recovery test were shown in Table 2.
Sample analysis
The established method has been successfully applied to the simultaneous determination of ten annonaceous acetogenins from five different Annona plant seeds. The contents (n=3) of ten annonaceous acetogenins were listed in Table 3. It can be seen that all ten compounds could be detected in all samples. The contents of these components varied in different Annona plantseeds. From the results, it was easy to note that bullatacin (3) was the most dominant compound in all samples. Its content ranged from 0.33 to 0.57 mg/g. Besides, the total content of the ten compounds ranged from 2.04 to 2.85mg/g which might be due to the differences in soils and climates in each region. Especially, the total content of the ten compounds in the Annona squamosa seeds was higher than other four Annona plant seeds. Thus it is necessary to control the main bioactive ACGs in different Annonaceae plant seeds by good agricultural practice (GAP). Then the quality of Annonaceae plant seeds could be assured.
|
|
|
| CONCLUSIONS |
|---|
A validated analytical method for qualification and quantification of annonaceous acetogenins from different Annona plantseeds has been developed, the new method was evaluated to be precise and accurate and successfully applied to determine the contents of ten major ACGs from five different Annona plant seeds. It was a convenient and precise method to assess the quality of different Annonaceae plant seeds.
|
|
ACKNOWLEDGEMENT |
|---|
The authors would like to acknowledge the financial support of the Natural Science Fund of Jiangsu Province.
|
|
REFERENCES |
|---|