| ORIGINAL ARTICLE |
|
|
Department of Pharmaceutical Chemistry, Faculty of Pharmacy, Cairo University, KasrEl-Aini St., Cairo 11562, Egypt
Corresponding Author: Bassam M. Ayoub, Department of Pharmaceutical Chemistry, Faculty of Pharmacy, Cairo University, Kasr El-Aini St., Cairo 11562, Egypt. Tel: +201225104337; Fax: +20224148452; E-mail: drbassamchemistry@hotmail.com.
| |
ABSTRACT |
| INTRODUCTION | |
|
|
EXPERIMENTAL |
|
|
RESULTS AND DISCUSSION |
|
|
CONCLUSION |
|
|
REFERENCES |
|
|
ABSTRACT
|
|---|
In this work, tworeversed-phase liquid chromatographic (RP-LC) methods have been developed for the determination of linagliptin (LNG) based on isocratic elutionusing a mobile phase consisting of potassium dihydrogen phosphate buffer pH (4.6)-acetonitrile(20:80,v/v) at a flow rate of 1 mL min−1. Two detection techniques have been applied either UV detection at 299 nm in the first method or fluorometric detection at 239 nm for excitation and 355 nm for emission in the second method. Chromatographic separation in the two methods was achieved on a Symmetry® cyanide column (150 mm×4.6 mm, 5μm). Linearity, accuracy and precision were found to be acceptable over the concentration ranges of 2.5-80 μgmL−1 for LNG in bulk and 2.5-15μg mL−1 for LNG in plasma with the first method and 5-160 μgmL−1 for LNG in bulk with the second method. The optimized methodswere validated and proved to be specific, robust and accurate for the quality control of the cited drug in its pharmaceutical preparation.
KEY WORDS: linagliptin; reversed-phase liquid chromatography;fluorometric detection; pharmaceutical preparation; plasma|
|
INTRODUCTION |
|---|
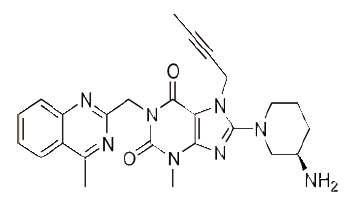
Linagliptin (LNG), 8-[(3R)-3-aminopiperidin-1-yl]-7-(but-2-yn-1-yl)-3- methyl-1-[(4-methylquinazolin-2-yl)methyl]-3,7-dihydro-1H-purine-2,6-dione] (Fig.1) is a novel hypoglycemic drug that belongs to dipeptidyl-peptidase-4 inhibitor class (1, 2).DPP-4 inhibitors represent a new therapeutic approach to the treatment of type 2 diabetes that functions to stimulate glucose-dependent insulin release and reduce glucagon levels. This is done through inhibition of the inactivation of incretins, particularly glucagon-like peptide-1 (GLP-1) and gastric inhibitory polypeptide (GIP), thereby improving glycemic control (3). Recently, DPP-4 inhibitors have been recommended in the treatment of diabetes mellitus to improve glycemic control (4) and it is effective in controlling the metabolic syndrome and resulted in significant weight loss, a reversal of insulin resistance, islet and adipocyte hypertrophy, and alleviated hepatic steatosis (5).
Only one method has been described for the determination of LNG in its pharmaceutical preparation based on reversed-phase liquid chromatography (6).
Due to the native fluorescence of LNG, our aim was to compare the two techniques of detection widely applied in routine analysis; namely UV and fluorometric detection and to try to develop a more sensitive method than that reported. Thus, we developedalternative LC methods for the determination ofLNG and applied it to the determination of LNG in plasma.In the first method (LC-UV), UV detection was applied for the determination of LNG in bulk, in plasma and in its dosage form. In the second method (LC-fluoro), LNG was determined in bulk and in its dosage form applying fluorometric detection based on the native fluorescence of the drug.
|
|
EXPERIMENTAL |
|---|
Instrumentation
The HPLC system consisted of a SchimadzuLC-20 AT Liquid Chromatograph (Japan) using a Symmetry® cyanide column (150 mm × 4.6 mm, 5 μm).The system was equipped with a flourometric detector (RF-551, Japan), UV-visible detector (SPD-20A, Japan) and an autosampler (SIL-20A, Schimadzu, Japan). An Elma S100 ultrasonic processor model KBK 4200 (Germany) was used.
Reagents and reference samples
Pharmaceutical grade LNG, certified to contain 99.80%, Tradjenta® tablets nominally containing 5 mg of LNG per tablet were supplied from Eli Lilly and company (USA). HPLC grade acetonitrile and methanol were purchased from Fisher Scientific (Loughborough, Leicestershire, UK). Potassium dihydrogen phosphate and orthophosphric acid (85%) were purchased from VWR Chemicals (Pool, England). Bi-distilled water was produced in-house(Aquatron WaterStill, A4000D,UK). Membrane filters 0.45 µm from Teknokroma (Barcelona, Spain) were used. All other chemicals and reagents used were of analytical grade unless indicated otherwise. Standard stock solutions of LNG (1 mgmL-1) were prepared by dissolving 100 mg of LNG in methanol in a 100 mL volumetric flask and completing to volume with methanol. The required concentrations were prepared by serial dilutions.
Plasma sample preparation
The spiked plasma samples of LNG were extracted after precipitation of proteins using 100 μL of perchloric acid (35% w/w). Then, the mixture was vortex-mixed and centrifuged (3 min). The supernatant was separated and transferred to another tube and a 25 μl volume was injected into the chromatograph.
Chromatographic conditions
Chromatographic separation was achieved on a Symmetry® cyanide column (150 mm × 4.6 mm, 5 μm) applying an isocratic elution based on potassium dihydrogen phosphate buffer pH (4.6) - acetonitrile (20:80, v/v) as a mobile phase. The buffer solution was filtered through 0.45µm membrane filter and degassed for 30 min in an ultrasonic bath prior to its use. The mobile phase was pumped through the column at a flow rate of 1 mL min-1. For LC-UV method, the UV detector was operated at 299 nm. For LC-fluoro method, the fluorometric detector was operated at 239 nm for excitation and 355 nm for emission. Analyses were performed at ambient temperature and the injection volume was 25 µL.
Sample preparation
Twenty tablets of Tradjenta® were weighed. An accuratelyweighed amount of the finely powdered Tradjenta® tabletsequivalent to 100 mg of LNG were separately made up to100 mL with methanol and sonicated to dissolve. The solutionswere filtered followed by serial dilutions to the required concentrations for each experiment.
Procedure
Linearity and repeatability.
Assay of LNG in bulk, plasma and Tradjenta® tablets. The procedure mentioned under LC-UV method in bulk was repeated using concentrations equivalent to 15-75 µg mL-1 LNG in bulk and equivalent to 4-12 µg mL-1 in plasma samples. The procedure mentioned under 2.6.1.2. was repeated using concentrations equivalent to 15-135 µg mL-1 LNG in bulk. For the determination of LNG in Tradjenta® tablets, the sample solution prepared under 2.6. was serially diluted and then injected in triplicates. The concentrations of LNG were calculated using calibration equation of each method.
|
|
RESULTS AND DISCUSSION |
|---|
HPLC greatly reduces the analysis time and allows for the determination of many individual components in a mixture using one single procedure (7). Due to the native fluorescence of LNG, we studied the two techniques of detection widely applied in routine analysis; namely UV and fluorometric detection. UV detection was selected and applied for the determination of LNG in plasma due to its more sensitivity and its applicability to the lower concentrations of LNG without distortions in the peak, also it gives linear reproducible results as shown in Table 2.
Methods development
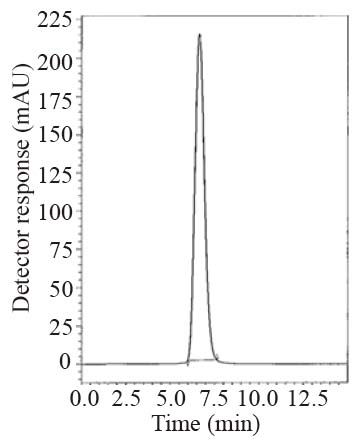
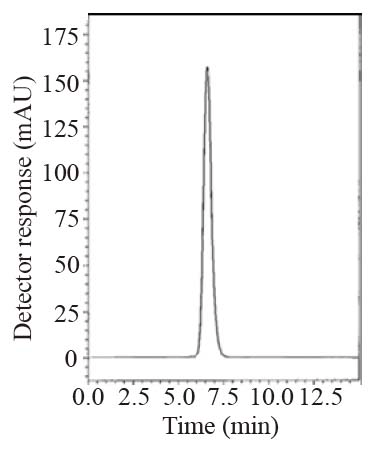
During the optimization cycle, various reversed-phase columns, isocratic mobile phase systems and different pH values of the buffer were attempted. Symmetry® cyanide column (150 mm × 4.6 mm, 5 μm) was found optimum. Various mobile phase compositions containing different ratios of organic and aqueous phases were tried in an isocratic mode. Acetonitrile was found optimum for the elution. Besides, different buffers at different pH values were attempted along with acetonitrile. Therefore, a mobile phase consisting of potassium dihydrogen phosphate buffer pH (4.6) - acetonitrile (20:80, v/v) pumped at a flow rate of 1.0 mL min-1, in an isocratic mode, gave good result. In LC-UV method, detection was carried out at 299 nm. In LC-Fluoro method, the fluorometric detector was operated at 239 nm for excitation and 355 nm for emission where high detector sensitivity was achieved at these wavelengths. The retention time was 6.6 min for LNG as in Fig. 2 and the retention time was 6.5 min for LNG in plasma as shown in Fig. 3.
System suitability tests
According to USP 2007 (8), system suitability tests are an integral part of liquid chromatographic methods in the course of optimizing the conditions of the proposed method. System suitability tests are used to verify that the resolution and reproducibility were adequate for the analysis performed. The parameters of these tests are column efficiency (number of theoretical plates), tailing of chromatographic peak and repeatability as %R.S.D of peak area for six injections and reproducibility of retention as %R.S.D of retention time. The results of these tests for the two proposed methods are listed in Table 1.
Methods validation
Linearity.
Accuracy.
Precision.
Specificity
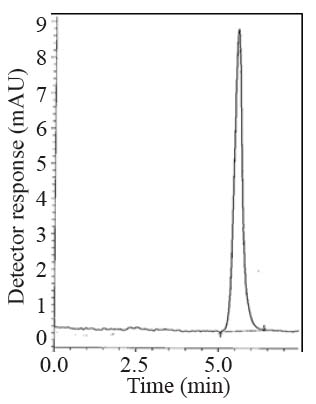
Specificity is the ability of the analytical method to measure the analyte response in the presence of interferences including degradation products and related substances. The chromatograms of the samples were checked for the appearance of any extra peaks. No chromatographic interference from any of the excipients was found at the retention times of the examined compounds (Fig. 2-4). In addition, the chromatogram of each compound in the sample solution was found identical to the chromatogram received by the standard solution at the wavelengths applied. These results demonstrate the absence of interference from other materials in the pharmaceutical formulations and therefore confirm the specificity of the proposed methods.
Limit of detection and limit of quantification
Limit of detection (LOD) which represents the concentration of analyte at S/N ratio of 3 and limit of quantification (LOQ) at which S/N is 10 were determined experimentally for the proposed methodsand results are given in Table 2.
Statistical analysis
A statistical analysis of the results obtained by the proposed method and the reference method was carried out by “SPSS statistical package version 11”. The significant difference between groups was tested by one way ANOVA (F-test) at p=0.05 as shown in Table 3. The test ascertained that there was no significant difference among the methods.
|
|
|
|
|
|
|
|
|
CONCLUSION |
|---|
The proposed LC methods proved to be simple, accurate and reproducible for the determination of LNG in reasonable run times. The methods were validated showing satisfactory data for all the method validation parameters tested. The developed method can be conveniently used by quality control laboratories.
|
|
REFERENCES |
|---|