| ORIGINAL ARTICLE |
|
|
La Sierra Academy, 4900 Golden Ave, Riverside, CA 92505, USA
Corresponding Author: David Han, La Sierra Academy, 4900 Golden Ave, Riverside, CA 92505, USA. E-mail: Davidhan1125@gmail.com.
Running title: ISOGENIC STEM CELLS BY CRISPR/CAS9 FOR HCM STUDY
| |
ABSTRACT |
| INTRODUCTION | |
|
|
CONCLUSION |
|
|
CONFLICT OF INTERESTS |
|
|
REFERENCES |
|
|
ABSTRACT
|
|---|
Hypertrophic cardiomyopathy (HCM) has been a heart disease causing the most sudden cardiac death among young people in US, and this problem still remains unsolved. In this paper, we will reintroduce HCM and discuss the methods that could be used to treat it. Many other studies, involving animal models; Induced pluripotent stem cells (iPSCs); CRISPR/Cas9, would also be mentioned to understand the disease better. Many techniques are useful in curing HCM. One of the helpful mechanisms would be iPSCs, which helps reprogramming a patient’s somatic cells, differentiating them into cells needed for the study. Due to different background issues, CRISPR/Cas 9 would then be added to generate isogenic iPSCs. Combining both mechanisms would help in comparing patient’s HCM iPSCs and control iPSCs, which provides us more information about pathogenic background of HCM and further assists in the process of curing HCM.
KEY WORDS: HCM; CRISPR/Cas9; iPSCs; heart disease and isogenic|
|
INTRODUCTION |
|---|
Hypertrophic cardiomyopathy (HCM) is a cardiac disorder characterized by left ventricular hypertrophy which predominantly occurs in interventricular septum (1). HCM is the most common genetic heart defect with prevalence of 1:500 in the general population, and its annual mortality rate is 1-5% (2). Over 400 autosomal mutations have been identified in genes encoding sarcomeric or sarcomere-related proteins in HCM patients (2). Despite much efforts in HCM research, studying the molecular mechanism underlying HCM has been hampered by a number of factors including difficulties to obtain human cardiac samples and propagate human heart tissue in cell culture systems. Although several murine HCM models have been generated and become a valuable tool in the disease study (3-5), they may not truly recapitulate human HCM as mouse heart is mainly constituted of α-myosin whereas the human heart mainly consists of β-myosin (6).
Induced pluripotent stem cells (iPSCs) reprogrammed from patient somatic cells have widely been used for disease modeling (7). In those studies, phenotypic differences observed in the iPSCs-derived cells of the patients and healthy individuals were usually claimed to be relevant to the disease pathophysiology. However, this traditional approach overlooks the impact of genetic background between healthy control and patients, which may result in misleading outcomes (8). Recently, isogenic iPSCs generated by CRISPR/CAS9 can eliminate the genetic background difference associated with the traditional IPSCs-based disease modeling, offering more reliable research outcomes (9, 10). In this article, I review the current methodologies in HCM studies and discuss the potential of isogenic iPSCs-based HCM disease modeling by using CRISPR/CAS9.
Genetic Causes of HCM
HCM was the first inherited cardiovascular disorder in which a genetic basis was identified. To date, over 400 autosomal mutations have been identified in at least 13 genes encoding sarcomeric or sarcomere-related proteins in HCM patients (Table 1). Of these genes, β-myosin heavy chain (β-MHC) and myosin-binding protein C (MyBP-C) are the two most common sarcomeric proteins that harbor approximately 70% of all mutations in HCM.
Recently, multiple mutations have been identified in approximately 5% of HCM patients (2). In contrast to HCM patients with a single heterozygous causative mutation, HCM patients with the multiple mutations are clinically more severe including earlier age of disease onset, more severe left ventricular hypertrophy, and more frequent and rapid progression to significant HCM complications of HCM (2). The severity of the phenotype is believed to directly correlate with the inherent protein dysfunction caused by the accumulation of multiple genetic mutations. The identification of multiple mutations in individual patients drastically changes genetic diagnosis, counseling, and treatment. Rather than single-gene testing, whole panels of genes should be tested in new families presenting with disease. Particularly, with multiple mutations, the likelihood of passing down the disease is even greater.
Methodologies in HCM studies
Over the past years, large achievements have been made in the management of HCM disease. However, definitive treatment to HCM is still lacking, and the molecular mechanism of HCM is poorly understood. Animal models and human iPS cells-based disease modeling have been widely used to study HCM molecular mechanism.
Animal Models
Genetically engineered animals have been very useful to study human HCM. Transgenic mice and rabbits that over-express mutant forms of myosin heavy chains exhibited the histopathological features seen in patients with enlarged left ventricles (11, 12). Similar studies of cardiac troponin-T and MyBP-C have been studied (4, 13, 14). As MyBP-C is one of main sarcomeric proteins implicated in HCM. Yang et al generated a mouse line that expresses a murine cardiac isoform of MyBP-C lacking both the titin and myosin binding sequences in attempt to mimic a class of MyBP-C HCM mutations (4). It was shown that the transgenically encoded, truncated MyBP-C protein is stable but not inserted efficiently into the sarcomere, resulting in a leftward shift in the pCa2+ force curve and a reduction of the power output (4). The researchers further noted that expression of the mutant MyBP-C protein leads to decreased levels of endogenous MyBP-C, resulting in a striking pattern of sarcomere disorganization and dysgenesis (4). Additionally, one of the most extensively studied mouse model of HCM was generated by introducing an Arg403Gln, a well characterized mutation in human HCM, into the α-cardiac myosin heavy chain gene. The heterozygous mouse genetically recapitulates the human HCM phenotype, and analysis showed features similar to the human disease (15).
In summary, the animal models of human HCM have demonstrated that a sarcomeric gene mutation is indeed the primary cause of HCM, and levels of mutant sarcomere protein expressed within the heart show some correlation with the severity of myocyte dysfunction. Although murine HCM models have become a valuable tool in the HCM disease study (3-5), they may not faithfully recapitulate human HCM. Indeed, several HCM studies have yielded conflicting results (16-20). For instance, it was suggested that mutant sarcomeric proteins impaired cardiac myocyte contractility, providing an impetus for compensatory hypertrophy (16, 20). In contrast, other studies found that various causative mutations produced inconsistent cardiac myocyte contractility, with some mutations reducing contractility and some mutations enhancing contractility (21, 22). In addition, some other molecular mechanisms have been proposed including perturbations in calcium cycling and sensitivity (23). Therefore, better approaches to elucidate the HCM molecular mechanisms are highly sought after.
Traditional iPSC-HCM Studies
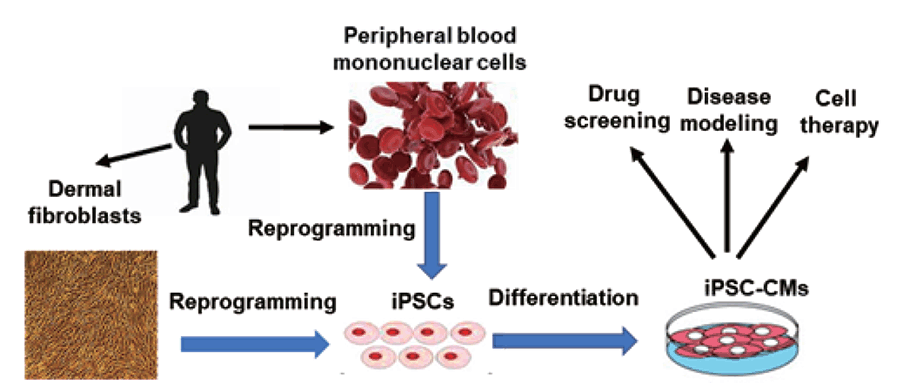
The recent development of iPSC reprogrammed from patient skin or blood cells, which are then differentiated into cardiomyocytes, can help our understanding of HCM disease and develop treatment strategies (23-29) (Figure 1). To dates, several iPSC-based HCM studies have been reported. In most of these reports, iPSC-derived cardiomyocytes are larger in size than those derived from healthy human iPSCs. For instance, Lan et al showed cellular enlargement in patient-specific iPSC-cardiomyocytes from a ten-member family cohort carrying a hereditary HCM missense mutation (Arg663His) in the MYH7 gene (23). In addition, Ojala et al studied two patients with HCM caused by Gln1061X mutation in myosin-binding protein C (MYBPC3) gene and Asp175Asn mutation in α-tropomyosin TPM1 gene (27). The cardiomyocytes derived from HCM patient-specific iPSC carrying either MYBPC3-Gln1061X or TPM1-Asp175Asn mutation displayed larger cellular size than those derived from healthy control iPSC (27). In addition, the myosin regulatory light chain (MYL2) mutation Arg58Gln is known to be associated with severe HCM, and cardiomyocytes derived iPSC of a HCM patient with MYL2-Arg58Gln were nearly 30% larger than the control iPSC-cardiomyocytes at day 60 (29). Other than larger cardiomyocyte cell size, myofibrillar disarray and abnormal electrophysiological properties were also observed in cardiomyocytes derived from HCM. For example, Han et al reported a HCM study by using iPSC reprogrammed from the dermal fibroblasts of a HCM patient with a single mutation (Arg442Gly) in the MYH7 gene (26). By Comparison to cardiomyocytes derived from healthy human iPSCs, they showed that the cardiomyocytes differentiated from the HCM patient iPSC displayed disorganized sarcomeres and irregularities in electrophysiology (26). Similar results were also observed in other studies. Zhou et al demonstrated that the percentage of myofibrillar disarray and cells with irregular beating in iPSC-cardiomyocytes of HCM patient with MYL2-Arg58Gln mutation was significantly higher than that in control cells (29). Moreover, Ca2+ plays a fundamental role in regulation of excitation-contraction coupling and electrophysiological signaling in the heart, and changes in Ca2+ handling was also seen in HCM iPSC-cardiomyocytes (23, 29).
Though iPSCs-based HCM modeling offer important insight in the understanding of disease mechanism. However, this traditional approach overlooks impacts of confounders such as genetic background, which may result in misleading outcomes. In the iPSCs-based dyskeratosis congenita disease modeling, for instance, two independent groups reported conflicting results with one showing regrowth of telomere and the other showing telomere decay (30, 31). In addition, Reinhardt et al. recently showed that neurons derived from iPSCs reprogrammed from different patients with Parkinson's disease could display distinct phenotypes. In particularly, one patient iPSC-derived neurons exhibited phenotypes similar to healthy control iPSCs-derived neurons (8). Such misleading results are mainly attributed to confounders of iPSC lines generated from patients and individual controls with different genetic background. Therefore, to ensure precise comparative analysis in iPSCs-based disease studies, it is imperative to eliminate the genetic and other variabilities between iPSC lines reprogrammed from patients and healthy individuals.
Isogenic IPSCs generated by CRISPR/Cas9 for HCM modeling
Recent rapid development in genomic editing technology CRISPR/Cas9 makes it feasible to generate isogenic iPSC lines which only differ in the disease causative mutations, would circumvent differences in genetic background and other variabilities among the cell lines. The CRISPR/Cas9 system is derived from a variation of prokaryotic defense which protects them from foreign genetic elements. CRISPR RNA (crRNA) and trans-activating crRNA (tracrRNA) come together to complex with Cas9 to identify and cut out these foreign sequences (32). A single-guide RNA (sgRNA) conjugated from the crRNA and tracrRNA can specifically introduce targeted double-stranded breaks (DSB) in human genome (32). The DSB then initiates cellular machinery to repair the DNA breaks typically by two repairing approaches, the non-homologous end-joining (NHEJ) repair which frequently results in nonspecific insertions and deletions (indels), and homology-directed repair (HDR) which utilizes DNA repair templates to generate knock-in of specific mutations (33).
There are two approaches to generate isogenic iPSC lines, correcting causative mutations in disease iPSCs or introducing disease causative mutations into health control iPSCs (Figure 2). Isogenic iPSC lines generated with either approach yield identical study outcomes, and recapitulate true disease mechanisms without genetic background variability (8, 34, 35). The first approach, to correct causative mutations in the disease-iPSCs, typically requires patient donor tissues for disease-iPSC reprogramming first. In addition, it is also associated with creation of multiple isogenic control lines with distinct genetic backgrounds when disease-iPSC lines of different patients are involved in a study, thus complicating isogenic iPSCs-based disease modeling (Figure 1). In contrast, the second approach, to directly introduce disease causative mutations into one control WT-iPSC line, is much simpler. It neither requires patient biopsies for iPSC reprogramming nor requires a different control line for a new disease-causative mutation (Figure 1). To date, there are only two studies of the isogenic iPSCs-based disease modeling have been reported for HCM disease (36, 37). Smith and colleagues reprogramed hiPSC lines from patients carrying the E99K mutation and a healthy non-carrier relative, and then generated isogenic iPSCs lines by correcting the mutation in the diseased iPSCs or introduce the mutation in the healthy non-carrier iPSCs using CRISPR/Cas9. Their study recapitulated HCM disease phenotypes including abnormal contractility, Ca2+ sensitivity/handling, arrhythmogenesis, and hypertrophic signaling (36).
|
|
|
|
|
CONCLUSION |
|---|
HCM, the most common genetic heart defect, is the leading cause of sudden cardiac death in young people in the United States. Current therapy can only relieve symptoms and prevent life-threatening complications, mainly due to unclear pathogenic mechanisms in HCM. Although animal models have offered some insight to HCM mechanisms, they may not faithfully recapitulate human HCM. In addition, all the traditional iPSCs-based HCM disease studies did not consider influence of the individuals’ genetic background difference and other variables which could result in misleading outcomes. This problem can be potentially overcome in isogeneic iPSCs generated by CRISPR/Cas9 which eliminate genetic background difference. Indeed, the recent two isogenic iPSCs-based HCM studies faithfully recapitalized HCM phenotypes (36, 37). Further comparative studies of isogenic disease HCM iPSC-CM with the control iPSC-CMs will help us understand the authentic pathogenic mechanisms underlying HCM as well as represent a valuable tool to screen the urgently needed drugs that can attenuate or reverse cardiac hypertrophy or fibrosis in HCM.
|
|
CONFLICT OF INTERESTS |
|---|
The authors declare that no conflicting interests exist.
|
|
|
REFERENCES |
|---|