| Review Article |
|
|
Laboratory of Hepatitis and Related Emerging Agents, Center for Biologics Evaluation and Research, US Food and Drug Administration
Corresponding authorDeborah R. Taylor, 8800 Rockville Pike, HFM310, Bethesda, MD 20892. Tel: (301) 827-3660; Fax: (301) 480-7928; E-mail: Deborah.Taylor@FDA.HHS.gov.
Running title: HCV cell culture systems
| |
ABSTRACT |
| INTRODUCTION | |
|
|
CONCLUSIONS |
|
|
ACKNOWLEDGMENTS |
|
|
REFERENCES |
|
|
ABSTRACT
|
|---|
Hepatitis C virus is a flavivirus that infects nearly 2% of the world population. There is no vaccine available and current therapy with interferon and ribavirin is expensive, not well tolerated and effective in only 60% of patients. HCV research has been hampered by the lack of a robust tissue culture system, but recent advances have made virus growth in culture possible. Here we review the current state-of-the-art and the molecular hurdles that have been met and those that still need to be overcome.
|
|
INTRODUCTION |
|---|
Hepatitis C virus (HCV) is an enveloped, positive-sense RNA virus of the Flaviviridae family that causes acute and chronic liver diseases (1). Six major genotypes have been identified for HCV that are further divided into numerous subtypes (2). The high mutation rate of the RNA genome generates viral diversity that leads to the existence of multiple quasispecies within an infected individual (3). About 170 million people are infected with HCV worldwide and these individuals are at high risk of developing cirrhosis and hepatocellular carcinoma.
Currently, there is no vaccine for HCV. Recombinant vaccine candidates have proved largely unsuccessful in mounting a protective response in Chimpanzees. Combination therapy utilizing interferon alpha (IFN-a) and ribavirin is successful in only half of the patients (4). A major obstacle in the development of effective vaccines and improved therapeutics has been the lack of a reproducible and efficient tissue culture system for propagation of HCV (5).
Genetic organization and life cycle of HCV
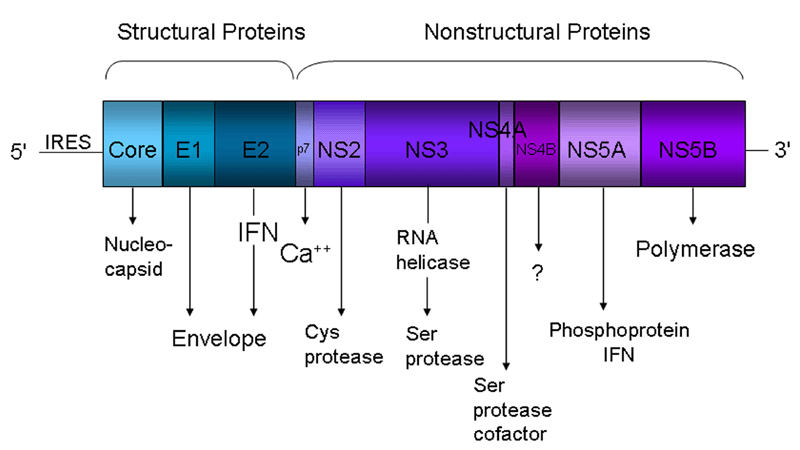
HCV is a small enveloped positive-strand RNA virus of the family Flaviviridae. The 9,600 nucleotides genome encodes a single large polyprotein that is post-translationally cleaved by viral and cellular proteases into 10 polypeptides; including three structural (core and the envelope glycoproteins E1 and E2) and seven nonstructural (NS) proteins (p7 and NS2, 3, 4A, 4B, NS5A and NS5B) (Figure 1). The structural proteins participate in viral entry and in the assembly of new particles, while the non-structural proteins play important roles in replication, assembly, and pathogenesis (6).
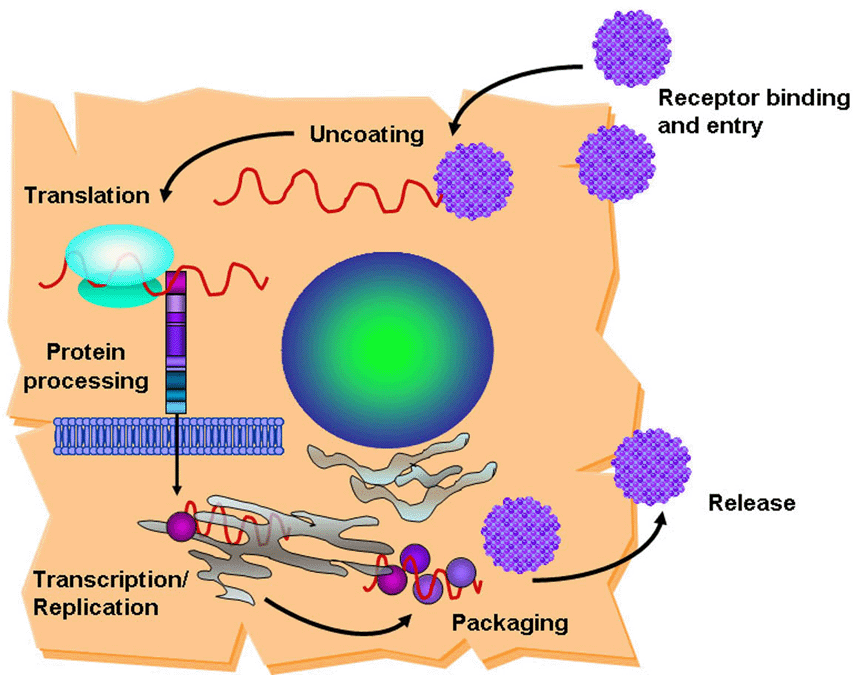
HCV is internalized in cells by receptor-mediated endocytosis. Several cellular co-receptors associated with lipid and lipoprotein metabolism have been proposed to mediate HCV entry into cells. CD81(7-9), the scavenger receptor class B type I (SR-BI) (10,11) and claudin-1 (12) are likely to be specific and it have been demonstrated as required for entry. Other cellular surface molecules have also been implicated in HCV entry: low density lipoprotein receptor (13,14), dendritic-cell-specific intercellular adhesion molecule 3-grabbing non-integrin (DC-SIGN) (15-17) and Liver/lymph node-specific intercellular adhesion molecule-3-grabbing integrin (L-SIGN) [(18,19)]. Nevertheless, cells expressing all of the putative co-receptors are still resistant to HCV, indicating the need of additional factors for viral entry (20). Binding of HCV to its receptor leads to fusion between the virion envelope and the cell membrane and release of the viral RNA into the cytoplasm of the host cell (Figure 2). The genome serves as a messenger RNA (mRNA) for translation of the viral proteins and constitutes the template for RNA replication. Translation depends on an internal ribosome entry site (IRES), located partially within the 5'-noncoding region (NCR), which binds to the host 40s ribosomal subunit without the need of most cell translation initiation factors [Figure 3; (21)]. The single viral polyprotein is cleaved by host signal peptidases and subsequently by viral proteases into the mature proteins. The HCV polymerase (NS5B) copies the positive-strand viral RNA into full-length anti-sense strand RNA, which becomes the template for both mRNA and genomic RNA. The full-length positive-sense RNA is packaged into the viral particles containing the core and envelope proteins. Virions presumably form by budding into the endoplasmic reticulum (ER) and are exported through the cell secretory pathway (22).
|
|
|
First attempts at HCV cell culture
Propagation of HCV has been extremely difficult. Before the identification of HCV as the causative agent behind non-A, non-B hepatitis infections, few infection systems were reported before 1999, and they all demonstrated low reproducibility and efficiency (5). These studies were initially performed using primary hepatocytes from humans or chimpanzees. Human fetal hepatocytes were successfully infected with HCV; however the system supported HCV replication that was only detectable by RT-PCR amplification (23). It was also shown that adult primary human hepatocytes could be infected and supported long-term culture for up to 4 months (24).
In addition, HCV was adapted to grow in the human T- and B-cell lines HPBMa10-2 and Daudi (25). Infectious virus was recovered from these cultures more than one year after infection. A model for chronic infection of suckling mouse brain (SMB) cells actively producing the virus was established by Deryabin et al. (26). They reported that persistent HCV contained in the SMB cultures induced a cytopathogenic effect in several cell lines, but these results have not be reproduced by other laboratories as of yet.
The replicon age: development of cell-based replication systems
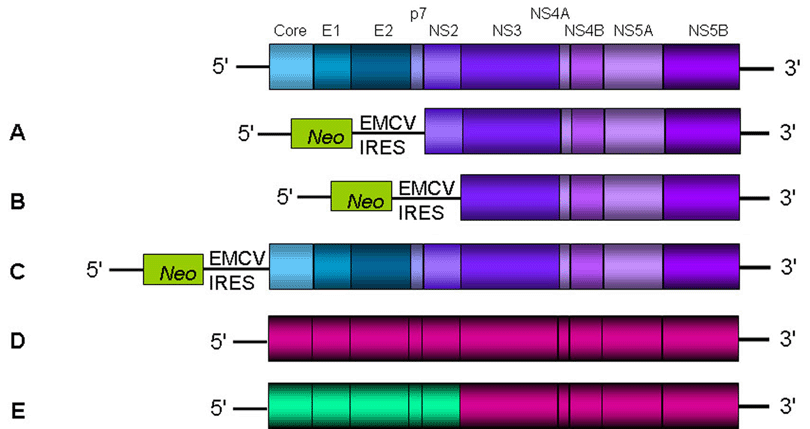
The development of HCV replicons in 1999 constituted a major advance and allowed the study of RNA replication and virus-cell interactions in the human hepatoma cell line Huh-7. The source of the first subgenomic HCV bicistronic replicon was total RNA isolated from a patient with chronic hepatitis C infection (27). Using long distance RT-PCR, the complete open reading frame was amplified in two overlapping fragments. After analysis of several clones of each fragment, an isolate-specific consensus sequence of genotype 1b was established. This replicon contains the HCV 5' noncoding region fused to the first 12 amino acids of the core protein, the neomycin phosphotransferase coding region (Neo), which confers resistance to the antibiotic geneticin, regulated by the internal ribosomal entry site (IRES) of HCV. The encephalomyocarditis virus (EMCV) IRES drives translation of the HCV non-structural proteins, and the HCV 3' noncoding region (Figure 3A, B). After transfection of Huh-7 cells with RNA transcribed in vitro by T7 RNA polymerase from the linearized replicon plasmid, cells were selected by culturing them in the presence of geneticin. These cells maintained the viral "subgenomes", but only a limited amount of geneticin-resistant colonies were obtained. Yet, these clones yielded high amounts of self-replicating HCV RNAs.
Subsequent studies using replicon containing cells, found a number of adaptive mutations in the HCV NS proteins, which increased RNA replication by several orders of magnitude (28-30). It was determined that through unknown mechanisms these mutations modify the activity of the viral replicase to enhance RNA replication in Huh-7 cells. Adaptive mutations were found primarily in NS3 (31,32), and in NS5A (33-35) .
Since then, several replicons have been established for different genotype 1b isolates (36,37), and for genotypes 1a (38) and 2a (39). Surprisingly, for reasons that are still under investigation, the genotype 2a replicon (JFH-1; derived from a Japanese patient with high levels of viremia and fulminant hepatitis) replicates approximately 20-fold more efficiently than the genotype 1-derived replicons without the need of cell culture-adaptive mutations (39,40) .
The host cell: improving HCV replication in highly permissive cell lines
Important progress has been made in the development of cell lines permissive for HCV growth. Treatment of replicon-containing Huh-7 cells with interferon alpha (IFN-α), resulted in the clearance of replicon RNA and selection of IFN-cured cells, which were designated Huh-7.5 (41). After reintroduction of the replicon, these cells supported HCV replication to much higher levels compared to that of the naïve parental Huh-7 cells. The mechanism is unknown, but several genetic lesions have been discovered in the Huh-7.5 cells (42). In addition, the level of CD81 cell surface expression in these cells was demonstrated to be a key determinant for productive viral entry (43). HCV RNA replication has now been achieved in HeLa, HEK 293, HepG2 and mouse hepatoma cell lines (44,45), indicating that viral replication is not restricted only to hepatocytes as it was believed for many years.
One more step ahead: development of full-length genomic HCV replicons
The isolation of highly permissive cell lines and the identification of adaptive mutations, constituted important tools for the establishment of efficient systems that could support autonomous replication of full-length genomic replicons. Yet, none of the RNAs generated from full-length replicons could produce infectious HCV virions (38,39,44). One possible explanation for this deficiency may be the interference of adaptive mutations with virus production. Mutations that promote efficient RNA replication in cell culture may be deleterious for later stages in the life cycle such as particle formation or release. This hypothesis is strongly supported by the fact that cell culture adaptive mutations present in the HCV FL-Con1 genome could block infectivity when inserted into a virus genome, preventing productive infection of chimpanzees (46). Additionally, the inability of the full-length genomic replicons to produce infectious virions could be due to the bicistronic nature of the system (Figure 3C). Possibly, the EMCV sequences, while driving up the efficiency of translation, may have a negative impact on packaging the virus.
Although the availability of the replicon system had made enormous contributions and provided extremely valuable tools to study viral replication, there are several concerns about the limitations of this system. First, the inability of the replicon to produce infectious virions; second, the requirement for adaptive mutations that are not found in naturally occurring isolates and last, the attenuated phenotype that these mutations showed in chimpanzees (46).
A surrogate system to study HCV infection
Major progress has been achieved with the generation of HCV pseudo-particles (HCVpp) (47,48), which are recombinant viral particles containing a retroviral core surrounded by an envelope, bearing native HCV glycoproteins E1 and E2. HCVpp are produced by co-transfecting cells with DNA plasmids containing a full-length HCV E1 and E2, the core proteins of either the human immunodeficiency virus (HIV) or murine leukemia virus (gag-pol genes), and a packaging-competent retroviral genome carrying a reporter gene. Transfected cells were capable of secreting approximately 105 assembled pseudoparticles per ml of culture supernatant (49). Successful entry into target cells can be monitored by measuring expression of the reporter gene. Various HCVpp have been developed with envelope proteins of genotypes 1a, 1b, 2a, 3a, 4a, 5a and 6a, allowing analysis of cross- and genotype-specific neutralization (50-60). It's been shown that HCVpp closely mimics the entry and serological properties of native and cell-culture produced HCV (49,51), and therefore they constitute an important tool for the study of virus attachment, receptor binding and fusion processes.
A breakthrough in HCV research: production of infectious viral particles
A key element towards the establishment of a robust system to study the entire HCV life cycle was the finding that transfection of a consensus JFH-1 genotype 2a full-length genome into Huh-7 cells rendered viral particles without the need of adaptive mutations (58) (Figure 3D). The secreted particles were infectious for Huh-7 cells and chimpanzees, and could be neutralized by CD81-specific antibodies and immunoglobulins from patients with chronic HCV infection. However, these particles showed a limited spread in naïve Huh-7 cells and consequently, the system rendered low viral titers. Continuous passage of JFH-1 transfected Huh-7.5.1 cells (a highly permissive Huh-7-derived cell clone, that was IFN cured from HCV replicons twice) allowed a substantial increase in the production of infectious particles, rendering 104-105 infectious units per ml of culture supernatant (60). Still the growth and spread of the JFH-1 virus was restricted to Huh-7-derived cell lines, since other cell lines like HepG2 and HeLa, failed to become infected.
A chimeric virus was constructed by inserting the core to NS2 region of genotype 2a HC-J6 into the JFH-1 replicon (55). Termed J6/JFH1, the virus was shown to release infectious particles from Huh-7.5 cells and produce relatively higher titers than JFH-1 (Figures 3F and 4). Subsequent studies demonstrated that chimeric J6/JFH1 viruses generated in vitro were infectious in chimpanzees and in a mouse model (61). Viruses recovered from these animals remained infectious in naïve Huh-7 cells. A series of chimeric genomes have since been constructed using the JFH-1 nonstructural region background, each allowing production of infectious viral particles of genotypes 1a, 1b, 2a, 3a and 4a (32,62-64).
|
In addition to the JFH1-bases infectious systems, several methodologies were applied to produce HCV infectious particles in cell culture. Stable human hepatoma cell lines containing a chromosomally integrated genotype 2a HCV cDNA that robustly produced HCV virions with up to 108 RNA copies per ml of culture media were established (65). Furthermore, Heller et al. (66) constructed an infectious genotype 1b cDNA between two ribozyme structures which were designed to generate authentic HCV RNA. These system supports production high levels of HCV virions.
Finally, a genotype 1a virus containing adaptive mutations, that was shown to infect Huh-7 cells in vitro (67) but to a lower infectivity than the JFH1 isolate, extends the current collection of tools available to study HCV.
|
|
CONCLUSIONS |
|---|
While all of these viruses replicate in culture, there are obvious limitations in these systems: a) they are restricted to hepatic-derived cell lines and b) only the JFH-1 isolate and JFH-1-derived viruses, robustly replicate and produce infectious virions, for reasons that are still under investigation. While JFH-1 came from a patient infected with genotype 2a virus, the reasons why this is the only strain of virus that grows in culture remains a mystery. Still needed is a cell culture system that can utilize virus from patient serum and not from a replicon or cDNA.
Although the available systems have several restrictions and their improvement will require further investigation, it is clear that HCV can now be efficiently produced and propagated in cell culture. These cell culture systems will provide the possibility to study every step of the viral life cycle, and will contribute to the development and evaluation of new antiviral strategies to control HCV infection.
|
|
ACKNOWLEDGMENTS |
|---|
We thank Ewan Plant and Maria Rios for comments on the manuscript. The findings and conclusions in this article have not been formally disseminated by the Food and Drug Administration and should not be construed to represent any Agency determination or policy.
|
|
REFERENCES |
|---|